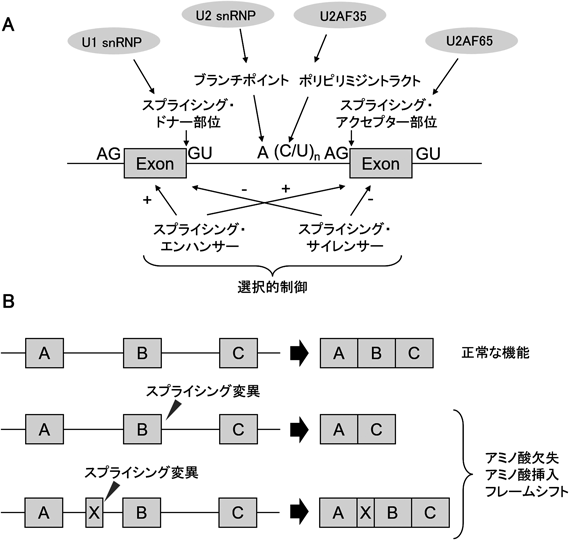
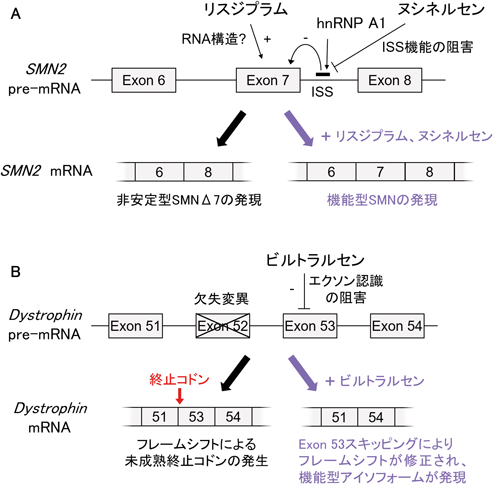
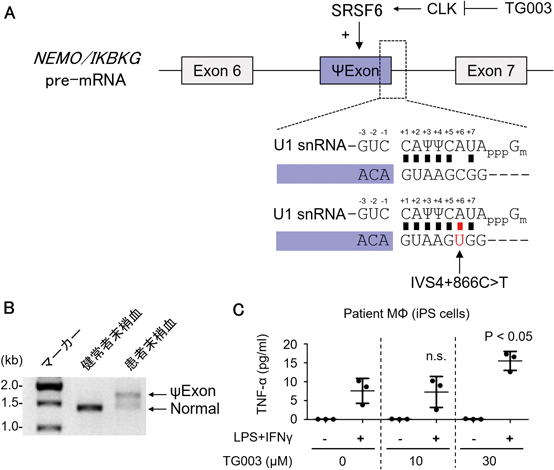
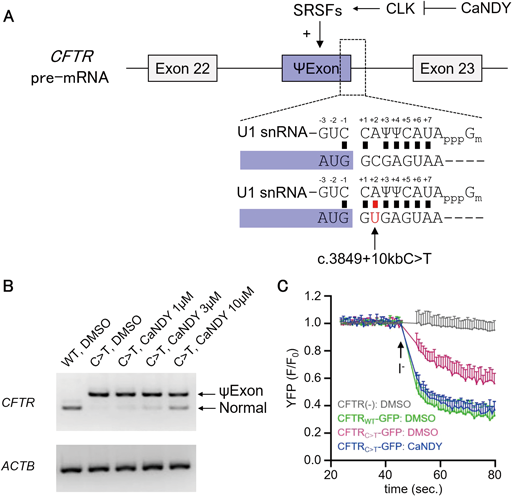
RNAスプライシング制御を標的とした創薬Drug development targeting aberrant RNA splicing
1 京都大学大学院医学研究科創薬医学講座Department of Drug Discovery Medicine, Kyoto University Graduate School of Medicine ◇ 〒606–8501 京都府京都市左京区吉田近衛町 医学部C棟3階 ◇ Building C, 3rd Floor, Yoshida Konoe Cho, Sakyo-ku, Kyoto 606–8501, Japan
2 京都大学大学院医学研究科形態形成機構学教室Department of Anatomy and Developmental Biology, Kyoto University Graduate School of Medicine ◇ 〒606–8501 京都府京都市左京区吉田近衛町 医学部C棟3階 ◇ Building C, 3rd Floor, Yoshida Konoe Cho, Sakyo-ku, Kyoto 606–8501, Japan
発行日:2022年12月25日Published: December 25, 2022