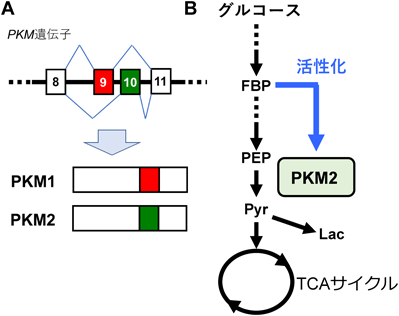
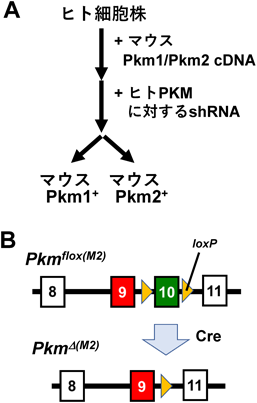
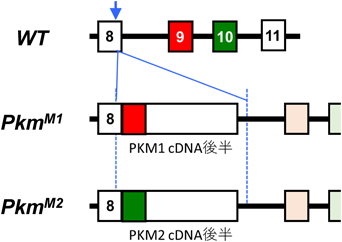
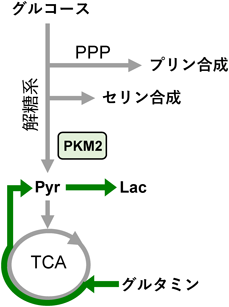
選択的スプライシングと,がんのワールブルグ効果Alternative splicing and the Warburg effect in cancer
1 宮城県立がんセンター研究所がん薬物療法研究部Division of Cancer Chemotherapy, Miyagi Cancer Center Research Institute ◇ 〒981–1293 宮城県名取市愛島塩手字47–1 ◇ 47–1 Noda-yama, Medeshima-Shiode, Natori 981–1293, Japan
2 東北大学大学院医学系研究科腫瘍生化学分野Biochemical Oncology, Tohoku University School of, Miyagi Medicine ◇ 〒980–8575 宮城県仙台市青葉区星陵町2–1 ◇ 2–1 Seiryo-machi, Aoba-ku, Sendai, Miyagi 980–8575, Japan
発行日:2022年12月25日Published: December 25, 2022