環境微粒子はマクロファージを刺激し炎症を惹起することが知られているが,その代表はシリカ(二酸化ケイ素)とアスベスト(石綿)であり,研究の歴史は古い.100年ほど前にはシリカ粉塵の曝露により肺炎や肺がんが発症することが知られており1),1960年代の肺線維化病変の電子顕微鏡解析によりシリカやアスベストは線維芽細胞よりもマクロファージに取り込まれていることが理解された.1980年代に入りシリカで刺激したマクロファージはinterleukin(IL)-1βを分泌することが報告された2).IL-1βは最も強力な炎症性サイトカインの一つとして知られる3).しかしIL-1βの産生機構は他のサイトカインの産生機構と比べて少し複雑であり,それが明らかになったのは最近(といっても20年前)のことである.
それはスイスのTschoppグループの生化学的手法により明らかにされた4).詳細は後述するが,活性化マクロファージ細胞質中ではpro-caspase-1, nucleotide-binding domain and leucine-rich repeat containing protein(NLRP:別名NALP),およびapoptosis-associated speck-like containing a CARD(ASC:別名Pycard)が複合体を形成してcaspase-1が活性化型となり,それがIL-1βの前駆体を切断して成熟型IL-1βができることが証明された4).このcaspase-1の活性化を起こすタンパク質複合体はインフラマソームと名づけられ,Toll-like receptors(TLRs)とは異なる炎症シグナルとして瞬く間に免疫学の最もホットな研究分野の一つとなり,それは今も続いている5–8).2008年に同じTschoppグループを含む複数の研究グループによる,シリカとアスベストはマクロファージNLRP3インフラマソームを活性化するという発見9, 10)を機に,無機微粒子による炎症と疾患の研究が注目されるようになった.マクロファージの炎症応答から慢性炎症性疾患の発症に至るまでには長期間にわたりさまざまな間質細胞や液性因子が複雑に関わるため,その病態についてはいまだに不明な点が多く残されている.本稿では,その初期応答,つまりマクロファージがどのように無機微粒子を認識して炎症を惹起するのかについて,我々の研究成果を含めて概説したい.マクロファージ細胞表面上には微生物を認識するための受容体が多く発現しているため,はじめにその特徴について簡単に説明する.
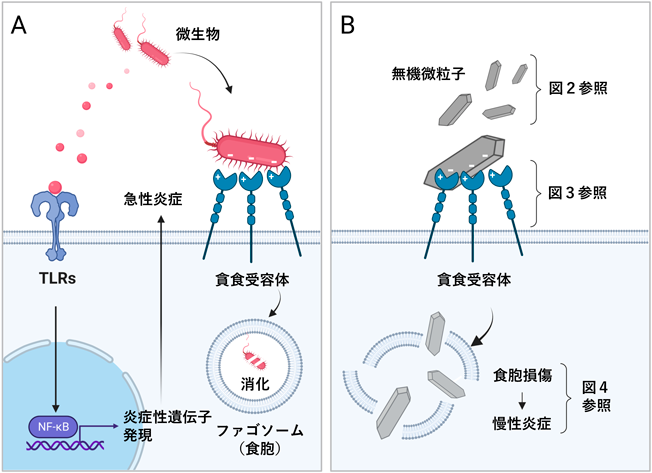
感染防御におけるマクロファージの役割は,生体内に侵入した病原微生物をいち早く感知して炎症を起こすことによってその情報を他の免疫細胞に知らせるとともに,病原微生物を貪食・消化して排除することである.我々の祖先は進化の過程で常に多種多様な微生物に曝露されてきたため,感染防御の砦として機能するマクロファージは微生物を認識する多くの受容体を獲得してきたと考えられる.マクロファージ細胞表面上の病原体認識受容体は大きく二つのタイプに分けられる(図1A).一つ目はサイトカイン受容体のように可溶性物質を高感度で認識する受容体であり,いわゆるセンサーとして機能する.その代表例としてTLRsがあげられる.TLRsは生体内に侵入してきた微生物から遊離してきたリポ多糖(LPS)やリポプロテインといった病原体関連分子パターン(pathogen-associated molecular patterns:PAMPs)を感知して二量体を形成し,Myd88などのアダプター分子を介して炎症シグナルを伝える11).しかしTLRsが直接微生物を貪食するわけではない.マクロファージが微生物を貪食した際にTLRsはファゴソーム(食胞)に集積するが12),それはおそらくPAMPsを探しに行っているためであり,TLRsがなくても貪食機能は低下しない13).二つ目のマクロファージ受容体は貪食受容体である.代表例としてスカベンジャー受容体ファミリー分子があげられる14, 15).多くのスカベンジャー受容体のリガンド認識部位には正に帯電するアミノ酸クラスターが存在し,また多くの微生物表面は負に帯電するため,両者は電荷依存的に結合する15).貪食受容体は多量体化して微生物を包み込む必要があるので,多数の分子がマクロファージ表面上に恒常的に発現している.このようにセンサーと貪食受容体が協調して微生物を排除する16).
無機微粒子に対しては貪食受容体のみが応答すると考えられる(図1B).ただし無機微粒子を特異的に認識する受容体が進化的に獲得されたとは考えにくいため,微生物の貪食受容体が無機微粒子を病原体と見誤って認識しているものと考えられる17, 18).無機微粒子は当然ファゴソーム内で消化されず,ファゴソーム損傷を引き起こして細胞死と炎症を惹起する19, 20).この消化されない無機微粒子はマクロファージ細胞死に伴い再び放出されて別のマクロファージに認識される.これが炎症を持続化する原因になると考えられる.またエンドトキシンなどのPAMPsが付着している環境微粒子に対してはTLRsが感知して炎症を惹起する.無機微粒子表面は貪食受容体のリガンドとなりうる特徴的な構造は持たず,概して均一な構造をとる.生体内で粒子表面にアルブミンやグロブリンといったタンパク質が吸着し,その層はprotein coronaと呼ばれる21)が,それがリガンドになっているとは考えにくい.多くの微粒子表面は微生物と同様にpH中性緩衝液中で負に帯電するため,電荷依存的に貪食受容体に認識されると考えられる.ただしすべてが電荷依存的認識で説明できるわけではなく,マクロファージは受容体を使い分けて無機微粒子を認識していると思われる.次にその認識機構と微粒子疾患について説明したい.
環境微粒子の研究はシリカとアスベストについて中心に進められてきたが,最近ではparticulate matter(PM)2.5(直径がおおむね2.5 µm以下の大気浮遊粒子状物質)やマイクロプラスチックも「持続可能な開発目標(SDGs)」の課題としてもあげられており,国際社会の関心は高い.また次世代材料として期待されているナノ粒子による健康被害が懸念され,今後の使用規制について国際的に激しく論争されている.
1)シリカと酸化チタン
シリカにはSiとOが規則性を持って配列する結晶(クリスタル)とその規則性がない非晶質(アモルファス)(図2)の二つの構造が存在する.シリカは地球上で最も多い化合物の一つであり,岩や砂の主成分であるためPM2.5,黄砂や多くの粉塵に含まれている22).結晶シリカ粉塵を大量に吸い込むと珪肺症と呼ばれる重篤な肺疾患を引き起こすことが知られるが22),最近の疫学研究から,シリカ粉塵の曝露レベルと肺がん,慢性閉塞性肺疾患(chronic obstructive pulmonary disease:COPD),リウマチ等のさまざまな炎症性疾患の発症との間に正の相関が認められている22).一方,非晶質シリカは安全であり,乾燥剤や賦形剤として多くの食品や医薬品に含まれている.しかし最近の動物実験から,非晶質でも粒子径が100 nm以下のナノ粒子については急性炎症を引き起こす可能性が指摘されている23, 24).酸化チタン(図2)も従来安全であると考えられ,ナノ粒子の原料としてシリカと並んで世界的生産量が最も多い.それは工業製品だけでなく食品の着色料や日焼け止めなどの化粧品に使用されてきた.しかしながらその一部のナノ粒子は動物実験で炎症を引き起こすことが報告されている25, 26).
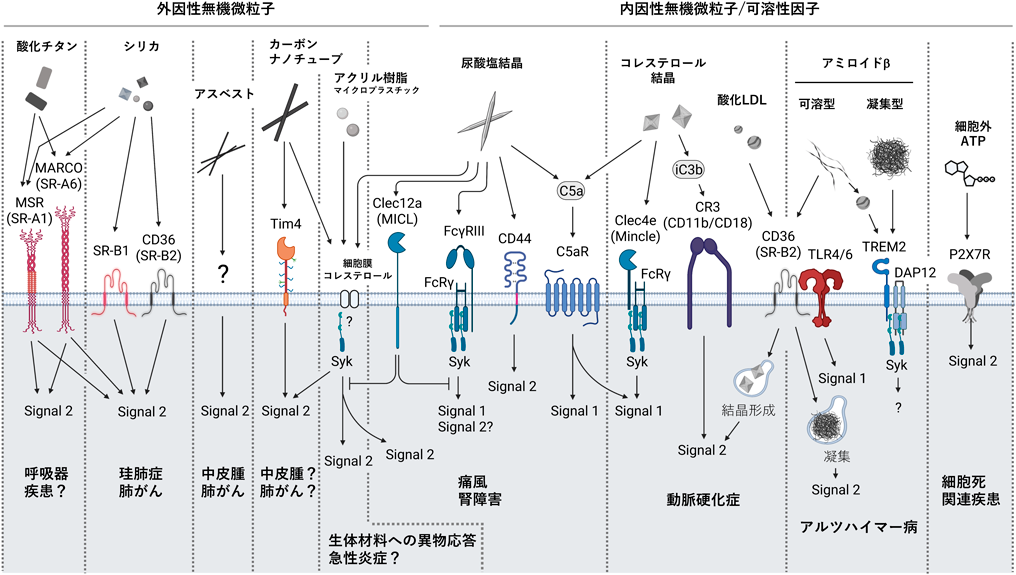
シリカと酸化チタンはいずれもscavenger receptor(SR)-A1(別名MSR1/CD204)およびmacrophage receptor with collagenous structure(MARCO:別名SR-A6)といったクラスAスカベンジャー受容体と電荷依存的に結合してマクロファージに取り込まれることが知られている17)(図3).理由は不明であるが,シリカは酸化チタンに比べてはるかに強いNLRP3インフラマソーム活性化と細胞死を誘導する27)(図4).またSR-A1とMARCOの遺伝子を欠損させてもその炎症応答は低下しない28, 29).そのため我々は発現クローニング法を行い,新たなシリカ受容体としてクラスBスカベンジャー受容体のSR-B1を同定した(本来はスカベンジャー受容体とはまったく構造の異なる予想外の分子がとれるのではないかと期待してスクリーニングをしたが,SR-B1しかとれなかった)27).SR-B1は肝細胞のHDL(高密度リポタンパク質)受容体として古くから知られている30).我々はSR-B1と同じファミリー分子のLIMP-2の結晶構造解析データを用いたホモロジーモデリングによりSR-B1の3次元構造を構築し,そのモデルからSR-B1も電荷依存的にシリカとHDLを認識することが判明した27).また同じクラスBスカベンジャー受容体のCD36(別名SR-B2)もシリカを認識し,四つのスカベンジャー受容体をすべてブロックすると貪食と炎症応答が顕著に低下することが判明した.ただしいずれもスカベンジャー受容体も細胞内領域は短く,機能的ドメインを持たないため15),貪食シグナルを伝える他の何らかの共受容体が存在する可能性が考えられる.
2)アスベストとカーボンナノチューブ
アスベスト(図2)は天然の鉱物繊維であり安価で耐熱性や防音性に優れ,“夢の素材”として1960年代の高度経済成長期に多くの建築材料に使用された.2030年ごろをピークに多くの老朽化したアスベスト含有建設物の解体作業が行われると見込まれ,その曝露による健康被害が懸念されている.アスベストにより誘発される中皮腫とは肺の外側に位置する胸膜の中皮細胞層に由来する悪性腫瘍であり,その曝露後20~30年経過して発症する31).そのため使用規制後50年以上経過した今もなお全国でアスベスト訴訟が起きている.
カーボンナノチューブ(carbon nanotubes:CNTs)はナノテクノロジーの代表的産物であり,電池,半導体,航空機,宇宙関連,医療といった多岐にわたる産業での次世代材料として期待されている32).しかしながら,一部の多層CNTs(multi-walled CNTs:MWCNTs)はアスベストに類似した針状構造をとり(図2),実際にアスベストと同じように肉芽腫や中皮腫を起こすことが多くの動物実験で報告され33),今後の使用について慎重派と推進派の研究者間で激しく議論されている34–37).
中皮腫の発症に至るまでに多様な間質細胞が複雑に関与しており,その機構はいまだに十分に理解されていないが,初期段階においてはアスベストあるいはMWCNTsを貪食したマクロファージ炎症応答が重要だと考えられる38).マクロファージによる認識機構については,微粒子に吸着した血清補体成分や免疫グロブリンなどの生体成分によるprotein coronaがリガンドになりうると考えられてきた39).しかし我々の実験では血清存在下でCNTsの補体受容体やFc受容体への結合が認められなかった40).またCNTsは疎水性相互作用により直接細胞膜リン脂質に結合して受容体非依存的に細胞内に取り込まれるとも考えられている41).もしこれら結晶が直接細胞膜に結合するのであれば,すべての細胞に同じように結合するはずである.しかしながら実際はマクロファージに最もよく結合する.この事実から我々はマクロファージ特異的な認識機構があると考えた.
そこで我々は,はじめにクラスAおよびクラスBスカベンジャー受容体について検討したが,どの受容体もCNTsに顕著に結合しなかった.次にマクロファージcDNAライブラリーを用いた発現クローニング法によりCNT受容体を探索したが,CNTsを取り込んだ細胞は死んでいくなどの理由でうまくいかなかった.その次にさまざまなマクロファージ受容体を個別にマウス線維芽細胞株NIH-3T3に導入し,順次CNT結合能を評価した.その結果,予想外にphosphatidylserine(PS)受容体のT cell immunoglobulin mucin(Tim)442)がCNTsを認識することが判明した40)(図3).分子動力学シミュレーションによりTim4は細胞外IgV領域のW119とF120で構成される芳香族アミノ酸クラスターを介してCNTsを認識することが予測され,実際にこれらをアラニン残基に置換するとその結合能は完全に消失した.この結合様式はTim4とPSの結合様式とは異なる.一般的に疎水性アミノ酸クラスターは水にふれにくいタンパク質構造内部や細胞膜貫通領域に存在し,それがタンパク質の立体構造の安定化に寄与している43).しかしながら生化学の基本原則に反してTim4は水とふれる表面上に疎水性アミノ酸クラスターを持つ.その理由は不明であるが,その部分がπ–π相互作用(二つの芳香環がコインを重ね合わせたような状態で安定化する作用)を介したCNT結合に必須である.実際にTim4はCNTsに限らずカーボンブラックナノ粒子などの炭素微粒子とも結合し,芳香環を持たないアスベストには結合しない40).Tim4もクラスAおよびクラスBスカベンジャー受容体と同様に細胞内に機能的ドメインを持たないため,微粒子結合が主な役割だと考えられる.貪食に共受容体が必要か否かは不明であるが,貪食されたCNTsがファゴソーム障害を起こしてNLRP3インフラマソーム活性化を起こす40)(図4).
3)PM2.5
PM2.5は,気管支や肺胞に到達して沈着すると考えられ,その健康被害が懸念されている.解剖学研究によりヒト肺組織に炭素微粒子やシリカ粒子が沈着していることが頻繁に観察されている44, 45).疫学調査からも動物実験からもPM2.5の大量曝露は肺がんや気管支喘息などの健康被害をもたらすことが懸念されている20).ただしPM2.5は採取場所や採取時で成分が大きく異なり,粒子表面にはさまざまな化学物質や微生物由来成分等が付着している.最近その粒子表層はprotein coronaとは区別してeco-coronaと命名されている21).当然エンドトキシンが付着していればTLR4を介して炎症を起こし,化学発がん物質が付着していれば発がんにつながる可能性がある.そのため付着物質と粒子の成分を総合的に理解する必要がある.粒子成分として炭素,ケイ素,金属イオンなどが含まれることからスカベンジャー受容体やTim4によって認識される可能性が考えられるが,まだよくわかっていない.
4)マイクロプラスチック
特に欧州ではマイクロプラスチックについて関心が高く,研究も進んでいることから,その環境汚染にとどまらず人体への影響も懸念されている46, 47).我々がマイクロプラスチックを大量に飲み込んだ魚を食べても糞便中に排泄されるだけのように考えられるが,最近,健常人の血液中や肝硬変患者から切除した肝中にマイクロプラスチックが検出されたと報告されている48, 49).ただし別の研究者から実験中のコンタミの可能性が疑われており50),データの解釈に注意が必要である.またポリスチレンビーズを海洋水族館の海水に浸して実験的にeco-corona21)をまとわせるとマクロファージへの取り込みが亢進することも報告されている51).体内に入ったマイクロプラスチックが炎症を引き起こすか否かは不明であるが,実験的にはポリスチレンやアクリル樹脂のpolymethyl methacrylate(PMMA)ビーズがマクロファージNLRP3インフラマソームの活性化を引き起こすことが報告されている52, 53).PMMAは生体材料としても使用されており,このマクロファージ応答は体内に埋め込んだ後で起きる炎症反応の原因の一つとして考えられる.取り込み機構としてポリスチレンビーズは電荷依存的にクラスAスカベンジャー受容体に結合することがよく知られており14),PMMAビーズは直接細胞膜コレステロールに結合して受容体非依存的経路を介することが報告されている52)(図3).
シリカやアスベストによるNLRP3インフラマソーム活性化が報告されたことを機に,体内で形成される結晶微粒子も同様の機構でIL-1βを分泌し,炎症性疾患の発症・増悪に関わっていることがわかってきた.
1)尿酸塩結晶(monosodium urate crystals:MSU)
血中尿酸値が高いと痛風の発症リスクが高まることは昔からよく知られていたが,その理由は長い間不明であった.Rockグループによって死細胞から放出される炎症性因子として尿酸が同定されたこと54)をヒントに,TschoppグループはMSUがNLRP3インフラマソームを活性化することを明らかにした55, 56).MSUの取り込み機構についてはShiグループから,PMMAと同様にMSUは細胞膜コレステロールへの直接結合に依存するといった受容体非依存的貪食モデルが提唱された57).しかしコレステロールはすべての細胞膜に存在するのにもかかわらずMSUは主にマクロファージに取り込まれるため,マクロファージ特異的な取り込み機構が存在すると考えられる.我々はMSU表面もシリカ表面と同様にpH中性緩衝液中で負に帯電することを観察したが,スカベンジャー受容体との結合は認められなかった27).他のマクロファージ受容体のFcγRIIIや補体受容体のC5aRに認識されることが報告されているが,これらの受容体が貪食に関わっているかどうかは明らかにされていない58, 59).最近CD44がMSUの貪食に関与することが報告されている60).また免疫抑制受容体として知られるC型レクチン受容体のClec12a(別名MICL)はMSUと結合するが,炎症抑制シグナルを伝達することも報告されている61)(図3).
2)コレステロール結晶
脂質異常症は動脈硬化や心筋梗塞の発症や自己免疫疾患の増悪に関与することが知られているが,それらの炎症性疾患にもNLRP3インフラマソームの活性化が関わっていることが数多く報告されている62, 63).初めの報告はLatzグループによるアテローム性動脈硬化症モデルマウスを用いたin vivo実験で,NLRP3遺伝子欠損によりその病態が軽減されることが観察され,in vitro実験でコレステロール結晶はマクロファージNLRP3インフラマソームの活性化を誘導することを明らかにした64).コレステロール結晶はMSUと同様に補体を活性化し,その活性化によって発現誘導された補体受容体のCR3(CD11bとCD18のヘテロ二量体)によってマクロファージ細胞内に取り込まれることが報告されている65).またヒトにおいてはC型レクチン受容体のClec4e(別名Mincle)がコレステロール結晶を認識し,Mincleと会合するFc受容体γ鎖を介して炎症シグナルを伝達することが報告されている66)(図3).
コレステロール結晶だけなく,酸化LDLがCD36を介してマクロファージに大量に取り込まれると,マクロファージ細胞内でコレステロール結晶が形成されてNLRP3インフラマソームの活性化が起きることも報告されている67).同じように樹状細胞内にコレステロールが蓄積すると樹状細胞のNLRP3インフラマソーム活性化を介した炎症性サイトカインの分泌や抗原提示能が亢進することも認められ,脂質異常症による自己免疫疾患の増悪のメカニズムを説明している68, 69)(図3).
3)アミロイドβ(Aβ)とタウ
これらの脳内蓄積はアルツハイマーおよび前頭側頭型認知症といった神経疾患の発症に深く関与していると考えられる.Aβペプチドが直鎖状に凝集したAβ線維あるいはタウタンパク質凝集体を貪食したマイクログリアは,無機微粒子を貪食したマクロファージと同様にNLRP3インフラマソームの活性化を介してIL-1β分泌を誘導することが報告されており70, 71),実際にこれら疾患モデルマウスおよび実際のヒトの病態においてNLRP3が関与していることが報告されている71, 72).それらの取り込み機構についていまだによくわかっていないが,クラスBスカベンジャー受容体のCD36は可溶型Aβを取り込み,その結果,細胞内に蓄積したAβが凝集してNLRP3インフラマソームを活性化することが報告されている67)(図3).また活性化マイクログリアから放出されたインフラマソーム複合体が核となってAβ沈着ができることも報告されている72).免疫受容体のTREM2のアルツハイマー病への関与も数多く報告されている73–75).TREM2がAβを直接的あるいは間接的に認識して取り込むが,それが病態の増悪因子としても抑制因子としても作用するといった相反する報告があり,混沌としている73–75).
5. 微粒子によるNLRP3インフラマソーム活性化と細胞死誘導機構
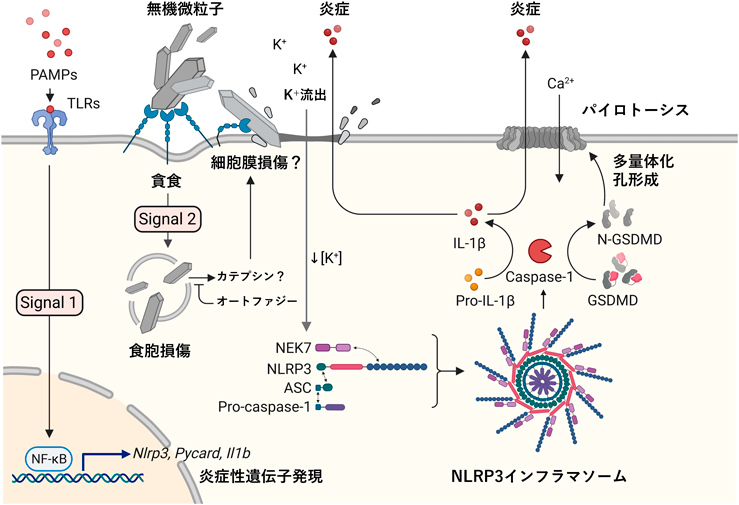
TNF-αやIL-6など多くの炎症性サイトカインは,翻訳されたタンパク質がそのまま活性化型として分泌されるが,IL-1βの分泌は厳重に管理されており,少なくとも二つのシグナルを必要とする5–8)(図4).シグナル1は,TLRs-NF-κB経路などを介したIL-1β前駆体の産生を誘導する.このとき細胞によってはNLRP3なども同時に誘導される.シグナル2は,NLRP3, ASC, pro-caspase-1を含むタンパク質複合体の形成を誘導する.これによって活性化されたcaspase-1がIL-1β前駆体を切断して成熟型IL-1βが分泌される.培養細胞を用いたin vitro実験では,微粒子を添加する前にシグナル1としてLPSなどの前処理が必要であるが,in vivo実験では必要でない.つまりシリカなどの微粒子をマウスに気管内投与すれば肺胞洗浄液中にIL-1βが分泌される.おそらく生体内では常在菌のPAMPsもしくは他の何らかの刺激によってIL-1β前駆体が発現していると思われる.
微粒子によるシグナル2の誘導にはファゴソーム損傷が関与していると考えられる18–20).たとえば非晶質シリカの場合,ナノサイズでもマイクロサイズでも同じようにSR-B1に認識され貪食されるが,ナノサイズだけがファゴソーム損傷を起こしIL-1β分泌を誘導する23, 27).カーボンナノマテリアルの場合,形状に関係なくTim4に認識されるが,針状構造のMWCNTsは激しくIL-1β分泌を誘導するのに対し,球状のカーボンブラックナノ粒子はまったく誘導しない40).またリソソーム膜を損傷する薬剤のL-Leucyl-L-Leucine methyl ester(LLOMe)はIL-1β分泌を誘導する76).このときオートファジーは損傷ファゴソームを除去することによって積極的にインフラマソームの活性化を抑制すると考えられる77)(図4).実際にMSU投与による腎障害モデルマウスにおいて腎特異的Atg5欠損により病態が増悪することが報告されている78).ただしファゴソームの損傷を起こす粒子と起こさない粒子を決定する物性はよくわかっていない.
ファゴソーム損傷から何らかのシグナルによって細胞膜損傷が起き,K+の細胞外流出が起きる.また細胞外アデノシン5′-三リン酸(adenosine 5′-triphosphate:ATP)(図3)は,ファゴソーム損傷は起こさないが,K+流出を起こす.細胞内K+濃度が低下することによって何らかを介してNEK7が活性化され,NLRP3インフラマソームが形成される79).活性化されたcaspase-1はIL-1β前駆体だけでなくパイロトーシス実行因子のgasdermin D(GSDMD)も切断する80).そのGSDMDにより細胞膜孔が形成され,さらにそれに続く小型膜タンパク質NINJ1が細胞膜破裂を誘導する81)(図4).GSDMDは細胞内グラム陰性菌感染などによる炎症には重要であるが81),微粒子による炎症には必須でないことが報告されている18, 82).その他の炎症性細胞死としてreceptor-interacting serine/threonine kinase-3(RIPK3)-mixed lineage kinase domain-like(MLKL)経路を介するnecroptosisがよく知られている83).上皮系細胞では微粒子による細胞死誘導はRIPK3-MLKL依存的であるが84),マクロファージでは非依存的であることが報告されている82, 85).微粒子が直接細胞膜を破壊する可能性86)も考えられるが,経時的な1細胞解析により微粒子による細胞死はプログラム化されているとも考えられ87),その詳細はわかっていない(図4).
in vitro実験では微粒子刺激によるIL-1β分泌はNLRP3に完全に依存するものの,モデルマウスを用いたin vivo実験では,微粒子疾患の病態にNLRP3はあまり重要でないこともわかってきた.たとえばアスベストによる中皮腫88),シリカによる珪肺89),そして高脂肪食摂取による動脈硬化症86)のいずれのマウスモデルにおいてもNLRP3遺伝子欠損とcaspase-1遺伝子欠損は病態を軽減しないことが明らかになっている.我々の予試験でもCNTsによる肉芽腫形成はNLRP3遺伝子欠損マウスと野生型マウスとの間に有意な差が認められなかった(著者ら未発表データ).これらの結果から,微粒子疾患の病態には,NLRP3-IL-1βよりも細胞死の方がより重要だと考えられる.さらに細胞死によって一度マクロファージに取り込まれた微粒子が放出されて,再び別のマクロファージに認識されるという負のスパイラルが,慢性炎症を起こすと考えられる.