1. はじめに~ユビキチンコードが制御する多彩な細胞内シグナル~
ユビキチンは,進化的に非常によく保存され,あらゆる細胞でユビキタスに発現する小さな球形タンパク質(約8.6 kDa)である.ユビキチンにより標識された細胞内の多くの不要タンパク質は,約2.5 MDaの巨大なタンパク質分解マシナリーであるプロテアソームに認識され,選択的に分解される(Aaron Ciechanover, Avram Hershko, Irwin Roseら3名は当該機構を発見し,タンパク質のターンオーバーという概念を確立した功績から2004年ノーベル化学賞を受賞した)1).タンパク質分解への関与から見いだされたユビキチン翻訳後修飾系ではあるが,現在ではシグナル伝達をはじめとする多彩な細胞内生理機能への関与が証明されており,細胞内システムを稼働させる基盤原理の一つと考えられる.
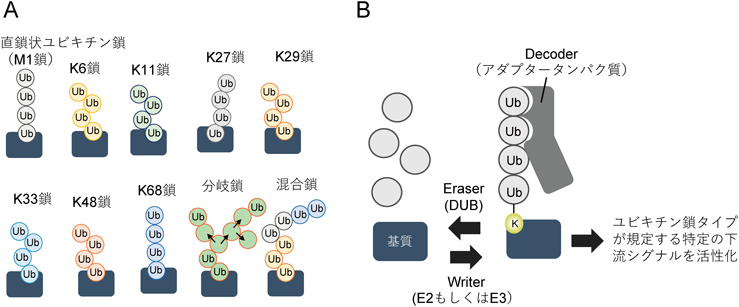
ユビキチン修飾系は特殊な生化学反応機構を有しており,エネルギーとしてATPを利用しつつ,3種類の酵素[ユビキチン活性化酵素(E1),ユビキチン結合酵素(E2),ユビキチンリガーゼ(E3)]が協同することで,特定の基質タンパク質の主にリシン残基へとユビキチンを付加する.他の翻訳後修飾機構とは異なり,ユビキチン修飾系は修飾体(基質タンパク質上のユビキチン)自体がタンパク質であるため,タンパク質に付加されたユビキチンがさらにユビキチンにより修飾されるという特徴を持つ.ユビキチンに対するこのような連続的な付加反応は,結果としてユビキチンどうしが数珠状に連結した,いわゆるユビキチン鎖を基質タンパク質へと結合させる(ポリユビキチン化)(図1A).さらに,不要となったユビキチン鎖を除去するため脱ユビキチン化酵素(deubiquitinase:DUB)が機能する仕組みも存在し(図1B),かつ,数百種類あるE3がそれぞれ基質選択性を有することで,細胞内のユビキチン修飾系の時空間的なON/OFFを厳密に制御することが可能となる.
ユビキチン修飾系が数多くの異なる細胞生理機能に関与可能な理由として,ユビキチン鎖自体が持つ構造多様性の存在があげられる.ユビキチン鎖の構造的相違は,ユビキチン結合ドメイン(ubiquitin-binding domain:UBD)を持つアダプタータンパク質の選別へとつながり,ユビキチン鎖へリクルートされたアダプタータンパク質は特定の下流シグナルの駆動を可能とする(図1B).モノユビキチン(基質に対して一つのユビキチンが結合)とポリユビキチンの違いはもちろん,ユビキチンどうしの結合様式に基づき形成される複数タイプのユビキチン鎖の存在が,構造上の多様性を拡張させている2).ユビキチンは七つのリシン残基を有するため,近位(基質タンパク質側)ユビキチンのリシン残基と遠位ユビキチンのC末端グリシンが結合することで7種類のユビキチン鎖(K6, K11, K27, K29, K33, K48, K63鎖)を形成することが知られており,さらには,直鎖状ユビキチン鎖(M1鎖)は,リシン残基の代わりにN末端メチオニンを介して連結することで新たなユビキチン鎖構造を作り出している(図1A)3).特にK48鎖とK63鎖は定常状態でも細胞内全ユビキチン鎖の大部分を占める典型的ユビキチン鎖に分類され,前述したタンパク質分解機構(K48鎖が主に関与)やシグナル伝達や膜輸送など(K63鎖が主に関与)細胞生存に重要な恒常的生理機能に利用されることがすでに明らかである.最近では,枝分かれしたユビキチン鎖からなる分岐鎖,同一鎖に異なるタイプのユビキチン鎖を含む混合鎖,ユビキチン自体のリン酸化・アセチル化修飾などの発見によりユビキチン鎖構造の理解がさらに複雑化している(図1A).ユビキチン修飾系の完全理解に向けて,ユビキチン鎖の高次構造情報に埋め込まれた“ユビキチンコード”と称されるいまだ謎の多い生命暗号の解明を目指し世界中で研究が進められている2).
本稿では筆者らが長年興味を持ち,研究対象としてきた直鎖状ユビキチン鎖に焦点を絞り,細胞内におけるその機能的役割から,臓器・個体維持における存在意義,病態生理への関与を中心に,筆者らの研究成果を含め解説する.直鎖状ユビキチン鎖は定常状態の細胞でほとんど生成されることのない非典型ユビキチン鎖の一つであり,多くの直鎖状ユビキチン鎖欠損細胞株は通常培養条件下で生存可能である4).一方で,直鎖状ユビキチン鎖の形成不全マウスは胎生致死に至ることが報告されており,直鎖状ユビキチン鎖形成が個体発生段階に必須である事実は大変興味深い点である5, 6).現在では,細胞外からの炎症因子[腫瘍壊死因子(tumor necrosis factor:TNF)やインターロイキン1β(interleukin-1β: IL-1β),Toll様受容体(Toll-like receptor:TLR)リガンドなど]の刺激に応答することで,細胞内での直鎖状ユビキチン鎖の形成が一過性に亢進することが知られている4, 7, 8).また,形成された直鎖状ユビキチン鎖は,NF-κB(nuclear factor-κB)やERK(extracellular signal-regulated kinase)シグナルの活性化を促進することに加え,デスリガンド依存的に誘導されるが外因性細胞死プロセスを強力に抑制する重要な生理機能を持つことが報告されており4),炎症環境条件下や免疫細胞との適切な細胞間コミュニケーションの成立に特異な役割を果たす.
前述したユビキチンコードの成立には,ユビキチン鎖を作り出すE2やE3(Writer),ユビキチン鎖と結合するUBDを持つアダプタータンパク質(Decoder),そしてユビキチン鎖を除去するDUB(Eraser)などが協同して機能することが知られており(図1B)2),次節では,直鎖状ユビキチン鎖のダイナミックな機能発現を促すこれら制御因子群の働きに着目する.また,世界的に最も理解が進むTNF誘導性NF-κBシグナルにおける機能解説にも主軸を置きながら,直鎖状ユビキチン鎖介在シグナルの生理学的必要性について論じていく.
2. 直鎖状ユビキチン鎖を形成する唯一のE3リガーゼ複合体LUBAC
ユビキチン鎖内の連結タイプの違いは,それらを作り出すWriterによって規定される.現在のところ,直鎖状ユビキチン鎖を形成可能な唯一のE3リガーゼとして,LUBAC(linear ubiquitin chain assembly complex)と呼ばれる複合体酵素が知られており,ほぼすべての細胞種においてユビキタスに発現している.LUBACはHOIP(HOIL-1L-interacting protein;120 kDa),HOIL-1L(heme-oxidized IRP2 ubiquitin ligase L;58 kDa)およびSHARPIN(SHANK-associated RH domain-interacting protein;40 kDa)の三つの因子により構成される複合体である3, 9).HOIPおよびHOIL-1Lは,ともにRBR[RING(really interesting new gene)-IBR(in-between-RING fingers)-RING]型ファミリーに属するE3リガーゼでC末端側にRBRドメインを持つが,直鎖状ユビキチン鎖形成はHOIPの活性中心を利用することが結晶構造解析とともに証明された10–13).HOIL-1LもしくはSHARPINの欠損が,他のLUBAC構成因子のタンパク質レベルでの量的減少を引き起こすことから,両者はLUBAC複合体の安定化に寄与する必須アクセサリー分子であると考えられる.
さらに,最近の我々の研究からHOIL-1LがLUBAC活性の自己調節機構に関与していることが明らかになった14).HOIL-1Lの示すわずかなリガーゼ活性に着目し解析を進めたところ,HOIL-1Lのリガーゼ機能の消失がLUBACの直鎖状ユビキチン鎖形成能を大幅に上昇させることを見いだした(結果として,後述する細胞・生体レベルでの細胞死保護作用を亢進させる).分子メカニズムの探索から,HOIL-1LによりLUBAC各構成因子がモノユビキチン化され,その後モノユビキチン上にHOIPが直鎖状ユビキチン鎖を付加する,いわゆるLUBACの自己直鎖化が過剰になることでLUBACが機能不全に陥る新たなLUBACの活性調節機構を提唱した.HOIPのN末端側には,直鎖状ユビキチン鎖を切断可能なDUBであるOTULIN(OUT deubiquitinase with linear linkage specificity)と結合する,またはアダプタータンパク質SPATA2(spermatogenesis associated 2)を介してCYLD(cylindromatosis)と結合するPUB(peptide:N-glycanase/UBA or UBX-containing proteins)ドメインを保持していることが知られている15).これらDUBが,LUBACに結合した直鎖状ユビキチン鎖を適度にトリミングすることでLUBACの正常な機能が保たれている.
LUBAC内では三者が1:1:1の割合で複合体を形成していると想定されているが,複合体全体の詳細な構造解析はいまだなされていない.また,ゲル濾過した細胞抽出液の解析からLUBAC複合体が約600 kDaほどの高分子量を持つことも示されており3),前述したDUBや新たな相互分子との会合状態も踏まえた細胞内での存在様式の解明が期待される.
LUBACが作り出す直鎖状ユビキチン鎖の細胞内生理機能に関しては,2009年に徳永・岩井ら(筆者の現所属研究室)により,TNFやIL-1β依存的なNF-κB経路の活性化に寄与することが初めて報告された(図2A)4).以降,特にTNFシグナルについては世界中で精力的に研究が進められてきた.NF-κB[RelA(p65),RelB, c-Rel, p105/p50(NF-κB1),p100/p52(NF-κB2)のホモまたはヘテロ二量体]は細胞の増殖や生存,分化,炎症応答など生体構築・維持に必須な転写因子群である.通常NF-κBは自身のRHD(Rel homology domain)を介してIκB(inhibitor κB)と会合し細胞質にとどまるが,ひとたびNF-κB活性化シグナルが入るとIκBはプロテアソームにより分解され,NF-κBの核内移行が可能となる.NF-κB経路の活性化経路は古典的経路[IKK1(IκB kinase 1)(IKKα),IKK2(IKKβ),NEMO(IKKγ)から構成されるIKK複合体が仲介する]と非古典的経路[IKK1とNIK(NF-κB-inducing kinase)が仲介する]に大別されるが,LUBACはNEMOと結合し,直鎖状ユビキチン鎖修飾を促すことで古典的NF-κB経路を活性化する.
前述したように直鎖状ユビキチン鎖は細胞外リガンドからの刺激により形成される(図2A).TNFとTNFR1受容体が結合すると,TNFR1細胞質側のDD(death domain)を介してTRADD(TNFR-associated death domain)やRIPK1(receptor interacting protein kinase 1)などアダプタータンパク質がリクルートされ,さらに会合するE3リガーゼTRAF2(TNF receptor-associated factor 2)やcIAP1/2(cellular inhibitor of apoptosis protein 1/2)の働きによりRIPK1上にK63やK11ユビキチン鎖が形成される(TNFR1 complex Iの形成)7).これらユビキチン鎖を足場として,TAK1-TAB1-TAB2/3複合体[TAB2/3のzinc finger(ZF)ドメインがUBD],IKK複合体(NEMOのZFドメインがUBD),LUBAC[HOIPのNZF1(Npl4 zinc finger 1)とSHARPINのNZFがUBD]がDecoderのもつUBDを介して局所的に集積する.このようにTNFR1細胞膜直下へと空間的に高密度に集積したIKK複合体やLUBACは,細胞染色時には斑点状に検出されるようであり16),おそらくこの局在変化により,LUBACは近傍のIKK複合体のNEMOやTNFR1 complex IのRIPK1を基質として認識することが可能になり,直鎖状ユビキチン鎖形成が促されると考えられる.結果として,NEMOやRIPK1上に付加された直鎖状ユビキチン鎖はHOIL-1LのNZFドメインを介してLUBACをさらに集積させ直鎖状ユビキチン鎖形成を促すことに加え,NEMOのUBANドメインを介してIKK複合体も集積させることでIKK複合体間におけるIKK2のトランス自己リン酸化(trans-autophosphorylation)を促し,IKK複合体の活性化を惹起する17).IKK複合体の活性化は,IκBのリン酸化とプロテアソームによる分解を誘導し,NF-κBの核内移行を促す.
このようにTNFR1を中心として構築されたシグナル複合体の形成は一過性であり,直鎖状ユビキチン鎖も形成後,数分ほどで減少に転じる.前述したOTULINやCYLDがEraser(DUB)として機能を果たしているためであり,これら直鎖状ユビキチン鎖形成を阻害するDUB因子群の消失はNF-κBシグナル活性化の異常延長を引き起こすことも観察されている.このように,直鎖状ユビキチン鎖に関与する一連の制御因子は,TNF刺激における古典的NF-κBシグナルの活性化と適切な炎症応答に必須であり(図2A),当該シグナルを正負どちらにも制御可能な機構を兼ね備えていることを考えると,サイトカイン応答シグナルのレオスタットとして中心的な役割を有していることも予想される.
LUBAC自体の翻訳後修飾もTNFシグナルの活性化に大きく影響する.TNF刺激依存的にTNFR1 complex Iへとリクルートされ,活性化したMST1(mammalian ste20-like kinase 1)は,HOIPの1066番目のセリン残基をリン酸化し,リガーゼ活性を低下させることでNF-κBシグナルを減弱させる18).逆にHOIPの784番目のリシン残基のユビキチン化やSHARPINの165番目のセリン残基のリン酸化は,リガーゼ活性に影響を与えずにLUBAC依存的なNF-κBシグナルの活性化を促しているようである19, 20).このように,LUBACが関与するTNFシグナルはさらに複雑な調節機構を備えている可能性があり今後の進展が期待される.
4. 直鎖状ユビキチン鎖が作り出す個体レベルでの生理的炎症寛容
1)慢性皮膚炎を発症するLUBAC欠損マウス
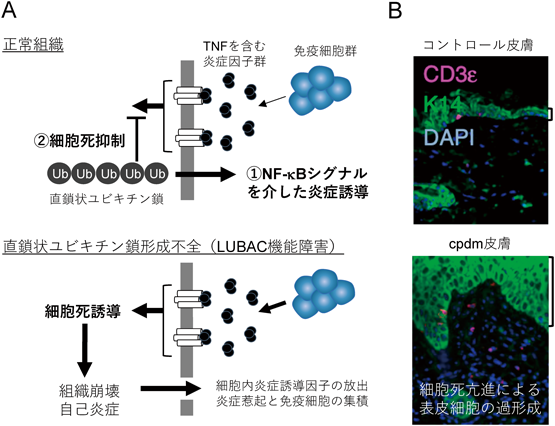
個体における直鎖状ユビキチン鎖形成の重要性については,遺伝学的にLUBAC構成分子を欠損させたマウスの解析からすでに明らかである.HOIPやHOIL-1L欠損マウスは,TNF-TNFR1シグナル依存的な血管内皮細胞の細胞死亢進に基づく血管形成障害によって,胎仔期中期embryonic day(E)10.5で致死に至る5, 6).これらの報告は,直鎖状ユビキチン鎖による特異な機能(デスリガンドであるTNF依存的な細胞死を抑制しつつ,適切なサイトカイン応答を促す)が正常な個体発生に必須であることを証明した.一方で,LUBAC構成因子であるSHARPINの欠損マウスは胎生致死には至らず,出生後3~5週間で慢性的な皮膚炎を発症することが知られている.このマウスは,SHARPINがLUBAC構成因子として同定される以前から,慢性皮膚炎を発症する一塩基欠損突然変異マウスとしてすでに報告されており,cpdm(chronic proliferative dermatitis mouse)と呼ばれている21, 22).cpdmの皮膚,特に表皮層では表皮の肥厚化,過角化やケラチノサイト細胞死の亢進が散見され,顆粒球(好中球や好酸球)やマクロファージなど炎症反応を促す自然免疫細胞の浸潤も認められる(図2B).また,顆粒球増加症(血中や免疫組織内(脾腫])や肝臓,肺を含むさまざまな臓器で炎症所見を呈する.筆者らはcpdmと皮膚特異的にSHARPINを強制発現するマウスを交配し,解析したところ,皮膚炎の改善とともに上記多臓器における炎症所見も検出されなくなったことから,cpdmで認められる多くの炎症所見は全身性慢性皮膚炎の二次的影響と考えられた23).HOIPやHOIL-1L欠損マウスとSHARPIN欠損マウスで明らかな表現型の違いが現れているが,その理由としてLUBACの量的損失,機能障害の程度の差によるところが大きい.マウス細胞を用いた実験では,LUBAC複合体安定化におけるHOIL-1Lの寄与は非常に大きいことがわかっており,HOIL-1L欠損はHOIPの存在量を著しく減少させる.一方でSHARPINを欠損させても残りの因子はわずかに減少するだけで,LUBAC機能の部分的減弱にとどまる.したがって,個体発生段階での影響はSHARPIN欠損ではほとんどないが,出生後の皮膚恒常性を維持するためには明らかにLUBAC機能が不足していると考えられた.個体においては,LUBACへの依存度が異なるさまざまな生理イベントが起こることがこれらの研究成果から十分理解できる.
2)外因性細胞死制御に基づいた炎症寛容機構
RelBやIκBα欠損マウス,Nemo+/−(X染色体上にコードされるため雌のみ)マウス,皮膚特異的IKK2欠損マウスなどで報告されているように,NF-κBシグナルの減弱はcpdmと同様に慢性皮膚炎を発症することが知られている.LUBACの機能により亢進したNF-κBシグナルはFLIP(FLICE-like inhibitory protein)やBcl-2(B-cell/CLL lymphoma-2)など抗アポトーシス因子群の発現誘導に重要であるため,cpdmについてもNF-κBシグナルの低下が皮膚構成細胞における細胞死誘発の原因の一つと考えられる.しかしながら,TNFで刺激した際に,NEMOやIKK2欠損細胞と比べて,LUBAC欠損細胞株では明らかに細胞死が亢進することを筆者らは観察しており,さらに個体レベルにおいてもHOIPやHOIL-1L欠損マウスが,IKK2, p65, NEMO欠損マウスと比べて早期に胎生致死となる事実から,LUBACがNF-κB転写誘導に依存した細胞死抑制機構とは明らかに異なる経路で炎症刺激から細胞を保護していることが想定される(図2A).
皮膚特異的なTNFR1欠損マウスとの交配により,cpdmの皮膚炎が明らかな改善を示すことから24),TNF-TNFR1シグナルがcpdmにおけるケラチノサイト細胞死誘導の主因と考えられる.近年,プログラム細胞死の多様性に関する研究が進み,TNF刺激によりCaspase 8依存的なアポトーシスとRIPK3-MLKL(mixed lineage kinase domain-like protein)依存的なネクロプトーシス(ネクローシス様表現型を呈するプログラム細胞死)が誘導されうることが明らかにされた25).Caspase 8のヘテロ欠損に加え,RIPK3を欠損させたcpdm(Caspase8+/−Ripk3−/−Sharpincpdm/cpdm)では完全に皮膚および多臓器の炎症所見が消失する(それぞれ片方の欠損では部分的な皮膚炎の改善を認める)ことから,LUBACはTNFR1 complex I内のRIPK1に直鎖状ユビキチン鎖を付加することで,この二つのプログラム細胞死を阻害していると考えられた24).RIPK1は,前述したようにTNF刺激依存的に,TRADDとともにTNFR1細胞内ドメインへリクルートされるアダプタータンパク質の一つであり,cIAP1/2, TRAF2およびLUBACの基質としてK11やK63,直鎖状ユビキチン鎖が付加される.LUBAC機能障害に基づいたRIPK1への直鎖状ユビキチン鎖形成不全のみならず,cIAP1/2阻害,CYLDやA20の機能亢進によるRIPK1上のK63鎖の除去が引き金となり,RIPK1がTNFR1 complex Iから離脱する.RIPK1は細胞質へと放出された後,細胞死の実行役を担うComplex II[Caspase 8, FADD(Fas-associated death domain protein),RIPK3とともに形成される複合体]を形成する.RIPK1に付加されたユビキチン鎖は,おそらくRIPK1のリン酸化状態を変化させることで細胞死抑制を達成するのかもしれない.TAK1阻害によってTNF刺激依存的なComplex IIの形成が促進することに加え,NEMOのUBANドメインを介してリクルートされるIKK複合体や,NEMOのアダプター分子であるTANK(TRAF family member-associated NF-κB activator)やNAP1(NAK-associated protein 1)を介してリクルートされるTBK1(TANK binding kinase 1)とIKKεによりRIPK1のリン酸化が促進され,Complex IIの形成を抑制することが報告されている26, 27).このように,LUBACおよびLUBACが形成する直鎖状ユビキチン鎖は,TNFシグナルにおいてNF-κBシグナル依存的・非依存的に強力な細胞死抑制機能を果たしており,cpdmが発症するような皮膚炎を恒常的に抑え込んでいる(図2).
さらに,cpdm皮膚炎を誘導する責任因子の探索研究から,TNF以外の外因性細胞死に関してもLUBACが抑制的に働いている可能性が報告されている.IL-1RAPもしくはIL-1R1受容体を欠損したcpdmの解析から,TNFR1欠損時ほどではないが,これら受容体欠損によりcpdmの皮膚炎が明らかに改善されることを観察しており,皮膚炎の増悪化にIL-1シグナルが寄与することが示された24, 28).また,IL-1R1と相同な細胞内領域を持つToll様受容体(TLR)ファミリーの多くでNF-κB活性化経路にLUBACが関与していることも以前より報告されており29, 30),LUBAC機能が欠損した表皮角化細胞株をTLR3のアゴニストであるpoly(I:C)で刺激するとTNFやIL-8などサイトカイン産生が著しく低下することに加え,刺激依存的かつTNF非依存的に細胞死を亢進させる31).TLR3が欠損したcpdmの作製から皮膚炎の明らかな改善が認められており,LUBACがTLRシグナル下流においても重要な細胞死抑制機能を果たしているようである31).IL-1R1/IL-1RAPやTLR下流シグナルに対するLUBACの関与は,これら受容体に共通する細胞内アダプタータンパク質であるMYD88(myeloid differentiation primary response 88)の欠損により,cpdm皮膚炎がほぼ完全に消失することからも証明された32).このMYD88欠損cpdmマウスの皮膚ではTNFの産生が減少していることが確認されており32),cpdmではIL-1βやTLRリガンドによる外因性細胞死亢進とともに,これら刺激により誘導されるTNFが皮膚炎の増悪化をさらに助長させる炎症機序が想定される.
細胞レベルでの生存維持にLUBAC欠失の影響はさほど認められない.他方で上記のように組織化・個体発生の各種イベントにLUBACが決定的な役目を果たすことがこれまで証明されてきた.これは外部刺激を受け,初めて形成・機能する直鎖状ユビキチン鎖の特徴によるものである.また,細胞外の炎症環境(TNFやIL-1βなど炎症性サイトカインが豊富な環境)に曝露された臓器・組織がその器質的・機能的恒常性を維持するには,TNFなどのデスリガンドによる細胞死誘導を回避しなければならず,直鎖状ユビキチン鎖の外因性細胞死抑制機能がその中心的な役割を担っていると考えられる(図2A).我々は,この外部炎症環境に応答して細胞内シグナル(NF-κBやERKシグナル)を亢進させつつ,同時に細胞死抑制機能を行使し組織崩壊を防ぐ,直鎖状ユビキチン鎖が果たすこの特殊な生理システムを「炎症寛容機構」と名づけた.炎症寛容機構は,炎症環境に対する組織側が発動する一種の適応機構と考えられる.
3)T細胞免疫正常化に基づいた炎症寛容機構
cpdmの皮膚炎症部位では獲得免疫システムの主体となるT細胞やB細胞などリンパ球の浸潤はほとんど認めない.また,RAG1(recombination activating gene 1)を欠失させ,T細胞やB細胞を消失させたcpdmで皮膚炎発症に影響がないことからも33),cpdm病態機序へのリンパ球の関与については長年にわたり注目されてこなかった.このような遺伝子変異に基づいた炎症性サイトカインの発現亢進や自然免疫システムの異常な活性化が引き金となり発症する全身性炎症性疾患は自己炎症性疾患(autoinflammatory diseases)と呼ばれ,リンパ球異常に基づく自己免疫疾患とは対照的な疾患と考えられている.しかしながら,筆者らはcpdmとLUBAC構成因子のコンディショナルノックアウトマウスを用いた詳細な解析から,前述した細胞死抑制機能に加え,LUBACおよび直鎖状ユビキチン鎖がT細胞免疫機能を維持することで同様に皮膚の恒常性獲得に貢献していることを発見しており,本項ではT細胞内でのLUBACの機能について説明しつつ,筆者らのこれらの成果について概説する.
最近,T細胞受容体[T cell receptor(TCR);T細胞が持つ膜タンパク質であり,抗原提示細胞などの膜上にある抗原ペプチド–MHC(major histocompatibility complex)複合体と会合することでT細胞の活性化を促す]のシグナル下流でLUBACが機能することが報告された34).TCRは抗原認識後,CD3分子群と複合体を形成し,細胞質領域CD3-ITAM(immunoreceptor tyrosine-based activation motif)内のチロシン残基がLck(lymphocyte cell-specific protein-tyrosine kinase)によってリン酸化され,さらにZAP70(zeta chain of T cell receptor associated protein kinase 70)などSH2ドメインを持つアダプタータンパク質が会合することで下流シグナルの活性化を引き起こす.TCRに依存したNF-κB経路の活性化には,細胞膜近傍へとリクルートされるPKCθ(protein kinase C theta)やシグナロソームCBM(CARMA1-BCl10-MALT1)複合体,その下流にIKK複合体の関与を必要とする.この経路を仲介する構成因子としてLUBACが新たに同定された34).CBM複合体は,スキャフォールドタンパク質であるCARMA1がPKCθによりリン酸化を受けて,不活性から活性モードへ構造変換されることで,他の構成因子とともに膜近傍へとリクルートされる.このとき,同時にLUBAC複合体もHOIPを介してCARMA1へ結合すると考えられる35, 36).筆者らの解析から,SHARPINやHOIPをそれぞれ欠損したハイブリドーマT細胞やJurkatヒトリンパ腫細胞株をTCR刺激した際に,NF-κB転写活性の低下に加え,NF-κBが誘導するサイトカインIL-2の産生も減少することが確認された.さらに,HOIP欠損時にはNF-κB転写活性が完全に消失する一方で,SHARPIN欠損時にはわずかな転写活性の低下を認めており,前述したSHARPIN欠失によるLUBACの不安定化と残存活性によるものであると考えられた.この結果は,抗原刺激時の十分なNF-κB活性化にはLUBAC構成因子がそれぞれ単量体としてではなく,LUBAC複合体として関与することを証明した23).HOIPとCARMA1の両者が直接結合するのか,それともHOIPのNZFドメインがBCL10上のK63ユビキチン鎖を認識して結合するのかいまだ統一見解が得られていない.また,CBM複合体のMALT1はTRAF6との結合能を有しており,TRAF6により形成されるCBM複合体上のK63ユビキチン鎖もLUBACを集積させる一因となる.その後,LUBACはBCL10をはじめとするCBM複合体構成因子に直鎖状ユビキチン鎖を付加することで35, 36),HOIL-1LのNZFドメインを介してLUBACのさらなる集積と,NEMOのUBANドメインを介してIKK複合体の集積・活性化を導き,最終的にNF-κBシグナル経路の活性化を可能にする.加えて,TCR–NF-κBシグナル経路におけるLUBACのリガーゼ活性非依存的な役割についても,我々の研究結果を含めその存在が示唆されており23, 34),LUBACが抗原刺激依存的なT細胞活性化を引き起こすメカニズムについてはさらなる解析が必要である.
TCR下流シグナルでのLUBACの役割が明らかになり,これまでcpdmにおいて見逃されていた個体レベルでの獲得免疫細胞(特にT細胞)へのLUBAC発現の意義についても注目されるようになってきた.2016年以降,複数のグループからSHARPINが欠失したcpdmで制御性T細胞[regulatory T cell(Treg);炎症抑制性のT細胞亜集団]が減少していることが報告された37).我々も同様の解析結果を得ており,さらにこれがT細胞における内因性の影響によるものか理解するために,Foxp3(Treg選択的に高発現する転写因子)プロモーター依存的にCreリコンビナーゼを発現するトランスジェニックマウスを用いて,Treg特異的なSHARPIN欠損マウスを作製し,解析を実施した23).このマウスはcpdmでの報告とは多少異なり,生後数週間はTregの細胞数はわずかに減少しているが,1~2か月ほどで野生型と同等の細胞数を示す(リンパ節内ではむしろ増加傾向).SHARPINが欠損したTregはTCR刺激依存的なNF-κBシグナルの減弱に加え,エフェクターTreg(高い炎症抑制機能を示すTregサブセット)が大幅に減少していた.さらに筆者らはこのマウスすべてが,生後10週前後にcpdmと同じような皮膚炎を発症することを見いだした.Lck-Creリコンビナーゼトランスジェニックマウスを用いることでTregを含むすべてのT細胞系列でSHARPINを欠損させると,皮膚炎になる確率が12%ほどまで低下した.これらの結果は,Treg特異的SHARPIN欠損マウスで発症する皮膚炎は炎症誘導性・細胞障害性T細胞の影響によるものであり,また,特にTregにおいては他よりもSHARPIN欠損で機能障害が生じやすいことが想定された.胸腺でのT細胞発生過程ではネガティブセレクションと呼ばれる機構が働き,自己抗原に対して高親和性のTCRを持つ未熟T細胞は,強いTCR刺激を受け取ることで細胞死が誘導され,発達過程で除去される(自己反応性のエフェクターT細胞の排除).一方で,自己抗原を認識し活性化するTregは発達過程で自己抗原からの強いTCR刺激を受け取る必要があることが知られている.SHARPIN欠損時のLUBAC残存活性では,おそらくTregの正常な発達やその後の末梢組織での機能発現には不十分であると考えられた(末梢Treg細胞数は生後時間を掛けて補充される).機能障害への影響は顕著であり,SHARPINを欠失したTregではT細胞活性化分子群に加え,Tregの炎症部位もしくは特定臓器への遊走を促すケモカイン受容体群Ccr4(CC chemokine receptor 4),Ccr6, Ccr9などの発現低下が認められた.Tregが皮膚組織指向性を持つためには前述したケモカイン受容体の発現が必要であるが,Treg特異的なSHARPIN欠損により,なぜ皮膚でのみ炎症が顕著に誘発されるのかそのメカニズムはいまだ謎である.末梢組織におけるT細胞免疫バランスはエフェクターT細胞とTregの両者の機能が拮抗する炎症制御モデルに依存することから,SHARPINはそのバランスを制御する重要因子であると考えられる.
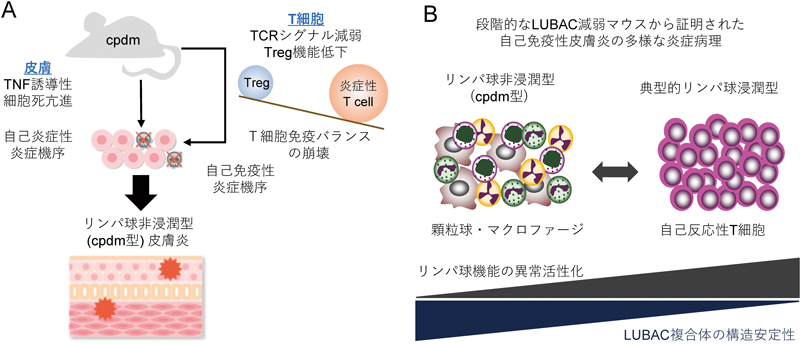
上記結果から,これまで想定されなかったTregの機能障害に基づく自己免疫機序がcpdmの皮膚炎発症に関与していることが十分に予想された.そこで次に我々は,コンディショナルトランスジェニックマウスを用いて,cpdmを遺伝子背景としたマウスでTregもしくはすべてのT細胞系統でSHARPINの発現を回復させた.予想どおり,これらのマウスではcpdm皮膚炎の明らかな改善を示し,さらに多臓器での炎症所見の消失,減少したTreg細胞数の回復(cpdmでのTreg細胞数の減少は,皮膚炎の二次的所見と結論づけた)を認めた23).以上の解析成果から,cpdmは細胞死易感受性がもたらす“自己炎症”だけでなく,同時にT細胞免疫バランスの崩壊がもたらす“自己免疫”の両者の機序を併せ持つ希有な炎症疾患モデルマウスであることが証明された(図3A).また同時に,直鎖状ユビキチン鎖を形成するLUBACが,免疫恒常性を保持するための主要要因として機能することを発見し,T細胞免疫正常化を介した皮膚の新たな炎症寛容機構を見いだした(図3A).以上,筆者なりの独自の研究プロセスから,複数の臓器で異なる機能を果たしながら皮膚恒常性維持を達成する,直鎖状ユビキチン鎖形成を基盤とした興味深い生理機構の一端を明らかとした.
5. 直鎖状ユビキチン鎖研究から見いだされた新たな自己免疫発症機序
本稿の内容から少し逸脱するが,筆者らの上記研究過程から自己免疫疾患の病態機序における新たな知見を見いだしたので紹介したい.Treg特異的なSHARPIN欠損マウスは皮膚炎を発症するが(前節参照),このマウスの皮膚炎症部位でリンパ球の浸潤は認められない(cpdmが自己炎症性皮膚炎と認識されていた事実に矛盾しない).それにもかかわらず,筆者らは前節で,このマウスでは自己免疫性(リンパ球依存的)の皮膚炎が起きていると記述した.筆者らはLUBACの複合体構造とこの複合体の安定性が構成因子に依存することに着目し,新たに2系統のコンディショナルノックアウトマウスSharpin−/−Hoip+/−とHoip−/−を作製し,Treg特異的かつ段階的にLUBAC機能を消失させたマウスモデルの比較解析を実施した(図3B)23).Treg内で段階的にLUBACを消失させることで,同一原因に基づき発症するが,重篤度の異なる自己免疫症状を作り出すことが可能となる.LUBAC残存活性の程度から,Treg機能はSharpin−/−>Sharpin−/−Hoip+/−>Hoip−/−(リガーゼ活性はゼロ)の順に低くなる.Sharpin−/−Hoip+/−は,Sharpin−/−と比べ明らかに早期に重篤な皮膚炎を発症し,またHoip−/−ではTreg内のLUBAC機能が完全に消失しており,重篤な全身性自己免疫症状ののち生後3週間前後で致死に至る.Treg特異的Hoip−/−の表現型はFoxp3欠損マウス(Scurfyマウスとして知られる)と酷似しており,典型的な自己免疫疾患モデルと考えられた.これら三者すべて皮膚炎を発症することから,免疫細胞に着目して病理学的比較を実施したところ,Sharpin−/−以外の系統では多数のT細胞浸潤を伴う自己免疫性の病理像を示し,対照的にSharpin−/−ではcpdmと同様にマクロファージや顆粒球など自然免疫細胞の浸潤が主に認められることを発見した(図3B).これら三者では同一要因によるT細胞依存的な炎症機序が想定されることから,リンパ球浸潤を認めないSharpin−/−での皮膚炎についても,非典型的な自己免疫性炎症所見を示しているものと考えられた.自己応答性T細胞の活性化程度の違いが,自己免疫疾患の異なる病理像を示す可能性を初めて証明した成果である(図3B).さらにTreg特異的SHARPIN欠損マウスの皮膚炎発症機序については,筆者らの詳細な解析から,活性化T細胞膜上に高発現する膜結合性TNFを介した皮膚表皮細胞の障害と,細胞死をトリガーとする自然免疫細胞の機能亢進が,結果として自己炎症様の皮膚炎を誘導する,新たな自己免疫疾患の炎症病態メカニズムを報告した23).
6. 直鎖状ユビキチン鎖依存的な炎症寛容機構の破綻とヒト炎症疾患
直鎖状ユビキチン鎖による適切な炎症誘導および外因性細胞死抑制(炎症寛容機構)が,インフルエンザウイルス感染時の肺胞上皮細胞やリポ多糖(lipopolysaccharide:LPS)投与による炎症性腸炎誘導時の腸管上皮細胞などで重要な働きをしていることが報告されており,マウス解析においては,特に各種上皮細胞が作り出す生体バリア機能と直鎖状ユビキチン鎖が密接に関与しているようである30, 38).肺や腸管上皮細胞においては,cpdmの皮膚のようにLUBAC機能低下により自然に慢性炎症が誘導されることはない.正常環境下ではこれらの臓器と皮膚との炎症性サイトカイン産生プロファイルが大きく異なる,もしくは細胞死に対する感受性の違いが考えられるが現在のところ明確な答えは出ていない.
近年の次世代シークエンサー技術による遺伝子解析技術の進歩により,ヒトにおいても,直鎖状ユビキチン鎖の形成不全や形成異常亢進に起因した自己炎症性疾患が複数報告されている.2012年にBoissonらによりHOIL-1L遺伝子変異症の患者が報告された39).両アレル遺伝子変異による発現・機能消失を招き,患者は周期性発熱と慢性的な炎症,細菌・ウイルス・真菌による反復性感染などを呈する.患者由来の線維芽細胞ではIL-1βに応答するNF-κBシグナルの活性化が減弱しており,ヒトにおいてもLUBAC機能が組織レベルでの炎症応答に必須であることを示す.一方で,線維芽細胞とは対称的に,単球ではIL-1β刺激による応答性の亢進を認めている.最近,樹状細胞(dendritic cell:DC)特異的にHOIPを欠損したマウスの解析から,LPS刺激によりHoip−/−DCの細胞死亢進とIL-1α/βの産生亢進が誘導されるとの報告もあり40),細胞種特異的な炎症亢進作用が種々の炎症所見を誘導している可能性が示唆される.また,マウスHOIL-1L欠損細胞で観察されるようにヒトでも一般的に外因性細胞死は亢進することが予想され,この細胞死に起因する病態機序も十分に考えられる.ヒトHOIP変異についてはこれまでに2例の報告があり41, 42),両アレルのHOIP遺伝子がL72Pミスセンス変異もしくは別の複合ヘテロ接合性変異によるもので,HOIL-1L遺伝子変異症の患者と同様の自己炎症と免疫不全症を呈する.こちらでも患者由来の線維芽細胞を用いた解析から,IL-1βやTNF-α刺激によるNF-κB活性化が減弱している一方,サイトカイン誘導性の細胞死の亢進が慢性炎症を引き起こす主因であると考えられる.
直鎖状ユビキチン鎖を切断するDUBであるヒトOTULINの変異症も報告されており,直鎖ユビキチン鎖過剰形成によるTNF誘導性の炎症異常亢進作用が自己炎症性疾患を引き起こす.OTULINの発現低下・機能消失を誘導する変異によって引き起こされる自己炎症性疾患はOTULIN関連自己炎症症候群(OTULIN-related autoinflammatory syndrome:ORAS)やOtulipeniaと呼ばれ,新生児期に好中球など白血球の増加を伴いながら周期性発熱や下痢,脂肪織炎,関節炎を呈するのが特徴とされる43–46).患者由来の線維芽細胞やB細胞を用いた実験では,正常人の細胞と比べ,直鎖状ユビキチン鎖の過剰形成が生じており,サイトカイン応答時のシグナルが亢進する47).多様な免疫細胞における炎症応答の異常亢進がORASの主因の一つではあるが,同時にLUBAC自体の自己直鎖状ユビキチン化が過剰亢進することでLUBAC機能が障害を受け,細胞死抑制機能が損なわれる現象も知られている48).LUBAC機能減弱に伴い生じる全身性の細胞死亢進が,前述したLUBAC欠損病と同様の症状を引き起こしている可能性もある.
これまで述べてきたように,LUBACの機能低下は細胞死亢進を招き,バリア機能の低下から全身性の慢性炎症へと移行することから,直鎖状ユビキチン鎖が関与する炎症寛容機構が,個体の恒常的生体環境を維持していることはいうまでもない.他方,直鎖状ユビキチン鎖形成が病気を作り出す状況についても考えてみたいと思う.先に述べたORASは,直鎖状ユビキチン鎖の切断不全により増強された炎症シグナルが原因となる慢性炎症疾患であり,直鎖状ユビキチン鎖依存的な炎症寛容機構が逆に炎症疾患を惹起する一例であるといえる.また,HOIL-1LによるLUBACの自己調節能については本稿2節で述べた.HOIL-1Lのリガーゼ活性を欠失させ,LUBAC(HOIP)のリガーゼ活性が異常に亢進したマウスでは,脾腫,多臓器への炎症細胞の浸潤,抗体産生亢進など軽微な免疫疾患様の所見も認めており,免疫関連疾患を惹起する素因としてLUBACの機能亢進の影響が強く疑われる14).現在までに,直鎖状ユビキチン鎖依存的なNF-κBシグナル活性化と,がんの発生や増殖,浸潤への関与を示唆する複数の報告が存在する49–54).筆者らは最近,成人で最も高頻度に発症する悪性リンパ腫であるびまん性大細胞型B細胞リンパ腫(DLBCL)の発症機序にLUBACの機能亢進が関与することを報告した55).DLBCLの中でも予後不良の活性化B細胞様DLBCL(ABC-DLBCL)の一部では,NF-κBシグナルの活性化を促すMYD88の変異とともに,LUBACの機能亢進をもたらす遺伝子多型が認められる56).MYD88活性型変異マウスはリンパ腫を発症するが,さらにLUBAC機能亢進を伴うマウスを作製すると,体細胞遺伝子変異の蓄積とともにリンパ腫の発症を促進した.体細胞遺伝子変異にはAID(activation-induced cytidine deaminase)が関与すると考えられ,通常AIDによるDNA障害は細胞死を誘導する.我々は,LUBACがこのDNA障害に基づく細胞死を抑制することでB細胞での遺伝子変異の蓄積を許容させ,リンパ腫の発症を促していることを示した55, 57).このようにがん形成とその悪性化にLUBACが関与している事実が次々と報告され続けているなかで,LUBACを標的とし,外因性細胞死抑制作用を解除することで,がん細胞の死滅を図るがん治療への応用研究も現在積極的に進められている.LUBAC阻害時にがん細胞を複数のサイトカインで併用処理すると細胞死誘導が明らかに亢進することが報告されており58, 59),そのメカニズムの解明やLUBAC阻害剤の開発,適応がん種の選定など今後の進展に期待したい.我々も現在,がんにおけるLUBACの興味深い振る舞いに注目しており,近くその詳細についても本誌にてご紹介できれば嬉しい限りである.
本稿では,ユビキチン鎖修飾系の中でも,外部炎症環境に依存して働く非典型直鎖状ユビキチン鎖修飾系に焦点を当て,筆者らの研究も含め,細胞から個体における炎症寛容機構の発見とその重要性について概説した.炎症環境においてはこれまで炎症性免疫細胞や炎症因子に焦点が置かれてきたが,本稿で解説したように,直鎖状ユビキチン鎖形成による組織側との適切な細胞間相互連携の成立が,正常な炎症プロセスの基盤にあることが理解できるようになってきた.炎症の場を制御するLUBACの存在は個体発生,炎症応答,がん形成など生体で起きる多彩なイベントに必須であると考えられ,今後もその生理・病態生理機能について新たな知見が蓄積されていくものと期待される.
謝辞Acknowledgments
本稿は,筆者が岩井一宏教授(京都大学大学院・医)の研究室に赴任した2014年以降の研究成果を中心に執筆させていただきました.本研究を長い間継続させていただき,また日々ご指導いただいた岩井教授に心から感謝申し上げます.また,これまでの私の研究人生を支えて下さった岩井研究室のメンバーの方々,共同研究者の方々,大学院時分よりお世話になっております田中啓二先生(東京都医学総合研究所),村田茂穂先生(東京大学),小松雅明先生(順天堂大学),その他多くの先生の方々につきましてもこの場を借りて感謝申し上げます.
引用文献References
1) Hershko, A. & Ciechanover, A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479.
2) Komander, D. & Rape, M. (2012) The ubiquitin code. Annu. Rev. Biochem., 81, 203–229.
3) Kirisako, T., Kamei, K., Murata, S., Kato, M., Fukumoto, H., Kanie, M., Sano, S., Tokunaga, F., Tanaka, K., & Iwai, K. (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J., 25, 4877–4887.
4) Tokunaga, F., Sakata, S., Saeki, Y., Satomi, Y., Kirisako, T., Kamei, K., Nakagawa, T., Kato, M., Murata, S., Yamaoka, S., et al. (2009) Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol., 11, 123–132.
5) Peltzer, N., Rieser, E., Taraborrelli, L., Draber, P., Darding, M., Pernaute, B., Shimizu, Y., Sarr, A., Draberova, H., Montinaro, A., et al. (2014) HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep., 9, 153–165.
6) Peltzer, N., Darding, M., Montinaro, A., Draber, P., Draberova, H., Kupka, S., Rieser, E., Fisher, A., Hutchinson, C., Taraborrelli, L., et al. (2018) LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature, 557, 112–117.
7) Haas, T.L., Emmerich, C.H., Gerlach, B., Schmukle, A.C., Cordier, S.M., Rieser, E., Feltham, R., Vince, J., Warnken, U., Wenger, T., et al. (2009) Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell, 36, 831–844.
8) Zinngrebe, J. & Walczak, H. (2017) TLRs Go Linear—On the Ubiquitin Edge. Trends Mol. Med., 23, 296–309.
9) Tokunaga, F., Nakagawa, T., Nakahara, M., Saeki, Y., Taniguchi, M., Sakata, S., Tanaka, K., Nakano, H., & Iwai, K. (2011) SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature, 471, 633–636.
10) Lechtenberg, B.C., Rajput, A., Sanishvili, R., Dobaczewska, M.K., Ware, C.F., Mace, P.D., & Riedl, S.J. (2016) Structure of a HOIP/E2~ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature, 529, 546–550.
11) Stieglitz, B., Rana, R.R., Koliopoulos, M.G., Morris-Davies, A.C., Schaeffer, V., Christodoulou, E., Howell, S., Brown, N.R., Dikic, I., & Rittinger, K. (2013) Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature, 503, 422–426.
12) Smit, J.J., Monteferrario, D., Noordermeer, S.M., van Dijk, W.J., van der Reijden, B.A., & Sixma, T.K. (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J., 31, 3833–3844.
13) Stieglitz, B., Morris-Davies, A.C., Koliopoulos, M.G., Christodoulou, E., & Rittinger, K. (2012) LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep., 13, 840–846.
14) Fuseya, Y., Fujita, H., Kim, M., Ohtake, F., Nishide, A., Sasaki, K., Saeki, Y., Tanaka, K., Takahashi, R., & Iwai, K. (2020) The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol., 22, 663–673.
15) Takiuchi, T., Nakagawa, T., Tamiya, H., Fujita, H., Sasaki, Y., Saeki, Y., Takeda, H., Sawasaki, T., Buchberger, A., Kimura, T., et al. (2014) Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells, 19, 254–272.
16) Tarantino, N., Tinevez, J.Y., Crowell, E.F., Boisson, B., Henriques, R., Mhlanga, M., Agou, F., Israel, A., & Laplantine, E. (2014) TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO-IKK supramolecular structures. J. Cell Biol., 204, 231–245.
17) Fujita, H., Rahighi, S., Akita, M., Kato, R., Sasaki, Y., Wakatsuki, S., & Iwai, K. (2014) Mechanism underlying IκB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol. Cell. Biol., 34, 1322–1335.
18) Lee, I.Y., Lim, J.M., Cho, H., Kim, E., Kim, Y., Oh, H.K., Yang, W.S., Roh, K.H., Park, H.W., Mo, J.S., et al. (2019) MST1 Negatively Regulates TNFα-Induced NF-κB Signaling through Modulating LUBAC Activity. Mol. Cell, 73, 1138–1149.e6.
19) Thys, A., Trillet, K., Rosinska, S., Gayraud, A., Douanne, T., Danger, Y., Renaud, C.C.N., Antigny, L., Lavigne, R., Pineau, C., et al. (2021) Serine 165 phosphorylation of SHARPIN regulates the activation of NF-κB. iScience, 24, 101939.
20) Fennell, L.M., Gomez Diaz, C., Deszcz, L., Kavirayani, A., Hoffmann, D., Yanagitani, K., Schleiffer, A., Mechtler, K., Hagelkruys, A., Penninger, J., et al. (2020) Site-specific ubiquitination of the E3 ligase HOIP regulates apoptosis and immune signaling. EMBO J., 39, e103303.
21) HogenEsch, H., Gijbels, M.J., Offerman, E., van Hooft, J., van Bekkum, D.W., & Zurcher, C. (1993) A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol., 143, 972–982.
22) Gijbels, M.J., Zurcher, C., Kraal, G., Elliott, G.R., HogenEsch, H., Schijff, G., Savelkoul, H.F., & Bruijnzeel, P.L. (1996) Pathogenesis of skin lesions in mice with chronic proliferative dermatitis (cpdm/cpdm). Am. J. Pathol., 148, 941–950.
23) Sasaki, K., Himeno, A., Nakagawa, T., Sasaki, Y., Kiyonari, H., & Iwai, K. (2019) Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat. Commun., 10, 3878.
24) Rickard, J.A., Anderton, H., Etemadi, N., Nachbur, U., Darding, M., Peltzer, N., Lalaoui, N., Lawlor, K.E., Vanyai, H., Hall, C., et al. (2014) TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife, 3, e03464.
25) Moriwaki, K. & Chan, F.K. (2017) The inflammatory signal adaptor RIPK3: Functions beyond necroptosis. Int. Rev. Cell Mol. Biol., 328, 253–275.
26) Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P.J., Giansanti, P., Heck, A.J., Dejardin, E., Vandenabeele, P., et al. (2015) NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell, 60, 63–76.
27) Lafont, E., Draber, P., Rieser, E., Reichert, M., Kupka, S., de Miguel, D., Draberova, H., von Massenhausen, A., Bhamra, A., Henderson, S., et al. (2018) TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol., 20, 1389–1399.
28) Liang, Y., Seymour, R.E., & Sundberg, J.P. (2011) Inhibition of NF-κB signaling retards eosinophilic dermatitis in SHARPIN-deficient mice. J. Invest. Dermatol., 131, 141–149.
29) Sasaki, Y. & Iwai, K. (2018) Crucial role of linear ubiquitin chain assembly complex-mediated inhibition of programmed cell death in TLR4-mediated B cell responses and B1b cell development. J. Immunol., 200, 3438–3449.
30) Sakamoto, Y., Sasaki, K., Omatsu, M., Hamada, K., Nakanishi, Y., Itatani, Y., Kawada, K., Obama, K., Seno, H., & Iwai, K. (2022) Differential involvement of LUBAC-mediated linear ubiquitination in intestinal epithelial cells and macrophages during intestinal inflammation. J. Pathol., 259, 304–317.
31) Zinngrebe, J., Rieser, E., Taraborrelli, L., Peltzer, N., Hartwig, T., Ren, H., Kovacs, I., Endres, C., Draber, P., Darding, M., et al. (2016) LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med., 213, 2671–2689.
32) Sharma, B.R., Karki, R., Lee, E., Zhu, Q., Gurung, P., & Kanneganti, T.D. (2019) Innate immune adaptor MyD88 deficiency prevents skin inflammation in SHARPIN-deficient mice. Cell Death Differ., 26, 741–750.
33) Potter, C.S., Wang, Z., Silva, K.A., Kennedy, V.E., Stearns, T.M., Burzenski, L., Shultz, L.D., Hogenesch, H., & Sundberg, J.P. (2014) Chronic proliferative dermatitis in Sharpin null mice: development of an autoinflammatory disease in the absence of B and T lymphocytes and IL4/IL13 signaling. PLoS One, 9, e85666.
34) Dubois, S.M., Alexia, C., Wu, Y., Leclair, H.M., Leveau, C., Schol, E., Fest, T., Tarte, K., Chen, Z.J., Gavard, J., et al. (2014) A catalytic-independent role for the LUBAC in NF-κB activation upon antigen receptor engagement and in lymphoma cells. Blood, 123, 2199–2203.
35) Yang, Y.K., Yang, C., Chan, W., Wang, Z., Deibel, K.E., & Pomerantz, J.L. (2016) Molecular determinants of scaffold-induced linear ubiquitinylation of B cell lymphoma/leukemia 10 (Bcl10) during T cell receptor and oncogenic caspase recruitment domain-containing protein 11 (CARD11) signaling. J. Biol. Chem., 291, 25921–25936.
36) Oikawa, D., Hatanaka, N., Suzuki, T., & Tokunaga, F. (2020) Cellular and mathematical analyses of LUBAC involvement in T Cell receptor-mediated NF-κB activation pathway. Front. Immunol., 11, 601926.
37) Redecke, V., Chaturvedi, V., Kuriakose, J., & Hacker, H. (2016) SHARPIN controls the development of regulatory T cells. Immunology, 148, 216–226.
38) Brazee, P.L., Morales-Nebreda, L., Magnani, N.D., Garcia, J.G., Misharin, A.V., Ridge, K.M., Budinger, G.R.S., Iwai, K., Dada, L.A., & Sznajder, J.I. (2020) Linear ubiquitin assembly complex regulates lung epithelial-driven responses during influenza infection. J. Clin. Invest., 130, 1301–1314.
39) Boisson, B., Laplantine, E., Prando, C., Giliani, S., Israelsson, E., Xu, Z., Abhyankar, A., Israel, L., Trevejo-Nunez, G., Bogunovic, D., et al. (2012) Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol., 13, 1178–1186.
40) Wu, X., Tang, Y., Zhang, S., Zhao, X., & Lin, X. (2021) MyD88-dependent signaling is required for HOIP deficiency-induced autoinflammation. J. Immunol., 207, 542–554.
41) Boisson, B., Laplantine, E., Dobbs, K., Cobat, A., Tarantino, N., Hazen, M., Lidov, H.G., Hopkins, G., Du, L., Belkadi, A., et al. (2015) Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med., 212, 939–951.
42) Oda, H., Beck, D.B., Kuehn, H.S., Sampaio Moura, N., Hoffmann, P., Ibarra, M., Stoddard, J., Tsai, W.L., Gutierrez-Cruz, G., Gadina, M., et al. (2019) Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front. Immunol., 10, 479.
43) Zinngrebe, J., Moepps, B., Monecke, T., Gierschik, P., Schlichtig, F., Barth, T.F.E., Strauss, G., Boldrin, E., Posovszky, C., Schulz, A., et al. (2022) Compound heterozygous variants in OTULIN are associated with fulminant atypical late-onset ORAS. EMBO Mol. Med., 14, e14901.
44) Damgaard, R.B., Walker, J.A., Marco-Casanova, P., Morgan, N.V., Titheradge, H.L., Elliott, P.R., McHale, D., Maher, E.R., McKenzie, A.N.J., & Komander, D. (2016) The deubiquitinase OTULIN Is an essential negative regulator of inflammation and autoimmunity. Cell, 166, 1215–1230.e20.
45) Damgaard, R.B., Jolin, H.E., Allison, M.E.D., Davies, S.E., Titheradge, H.L., McKenzie, A.N.J., & Komander, D. (2020) OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ., 27, 1457–1474.
46) Damgaard, R.B., Elliott, P.R., Swatek, K.N., Maher, E.R., Stepensky, P., Elpeleg, O., Komander, D., & Berkun, Y. (2019) OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med., 11, e9324.
47) Zhou, Q., Yu, X., Demirkaya, E., Deuitch, N., Stone, D., Tsai, W.L., Kuehn, H.S., Wang, H., Yang, D., Park, Y.H., et al. (2016) Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA, 113, 10127–10132.
48) Heger, K., Wickliffe, K.E., Ndoja, A., Zhang, J., Murthy, A., Dugger, D.L., Maltzman, A., de Sousa, E.M.F., Hung, J., Zeng, Y., et al. (2018) OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature, 559, 120–124.
49) Tomonaga, M., Hashimoto, N., Tokunaga, F., Onishi, M., Myoui, A., Yoshikawa, H., & Iwai, K. (2012) Activation of NF-κB by linear ubiquitin chain assembly complex contributes to lung metastasis of osteosarcoma cells. Int. J. Oncol., 40, 409–417.
50) Kharman-Biz, A., Gao, H., Ghiasvand, R., Haldosen, L.A., & Zendehdel, K. (2018) Expression of the three components of linear ubiquitin assembly complex in breast cancer. PLoS One, 13, e0197183.
51) Ruiz, E.J., Diefenbacher, M.E., Nelson, J.K., Sancho, R., Pucci, F., Chakraborty, A., Moreno, P., Annibaldi, A., Liccardi, G., Encheva, V., et al. (2019) LUBAC determines chemotherapy resistance in squamous cell lung cancer. J. Exp. Med., 216, 450–465.
52) Song, Z., Wei, W., Xiao, W., Al-Saleem, E.D., Nejati, R., Chen, L., Yin, J., Fabrizio, J., Petrus, M.N., Waldmann, T.A., et al. (2020) Essential role of the linear ubiquitin chain assembly complex and TAK1 kinase in A20 mutant Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA, 117, 28980–28991.
53) Song, K., Cai, X., Dong, Y., Wu, H., Wei, Y., Shankavaram, U.T., Cui, K., Lee, Y., Zhu, B., Bhattacharjee, S., et al. (2021) Epsins 1 and 2 promote NEMO linear ubiquitination via LUBAC to drive breast cancer development. J. Clin. Invest., 131, e129374.
54) Hua, F., Hao, W., Wang, L., & Li, S. (2021) Linear ubiquitination mediates EGFR-induced NF-κB pathway and tumor development. Int. J. Mol. Sci., 22, 11875.
55) Jo, T., Nishikori, M., Kogure, Y., Arima, H., Sasaki, K., Sasaki, Y., Nakagawa, T., Iwai, F., Momose, S., Shiraishi, A., et al. (2020) LUBAC accelerates B-cell lymphomagenesis by conferring resistance to genotoxic stress on B cells. Blood, 136, 684–697.
56) Yang, Y., Schmitz, R., Mitala, J., Whiting, A., Xiao, W., Ceribelli, M., Wright, G.W., Zhao, H., Yang, Y., Xu, W., et al. (2014) Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov., 4, 480–493.
57) Niu, J., Shi, Y., Iwai, K., & Wu, Z.H. (2011) LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J., 30, 3741–3753.
58) Freeman, A.J., Vervoort, S.J., Michie, J., Ramsbottom, K.M., Silke, J., Kearney, C.J., & Oliaro, J. (2021) HOIP limits anti-tumor immunity by protecting against combined TNF and IFN-γ-induced apoptosis. EMBO Rep., 22, e53391.
59) Zhang, Z., Kong, X., Ligtenberg, M.A., van Hal-van Veen, S.E., Visser, N.L., de Bruijn, B., Stecker, K., van der Helm, P.W., Kuilman, T., Hoefsmit, E.P., et al. (2022) RNF31 inhibition sensitizes tumors to bystander killing by innate and adaptive immune cells. Cell Rep. Med., 3, 100655.
著者紹介Author Profile
 佐々木 克博(ささき かつひろ)
佐々木 克博(ささき かつひろ)京都大学大学院医学研究科細胞機能制御学 講師.博士(生命科学).
略歴2006年東京理科大学薬学部卒業,11年東京大学大学院新領域創成科学研究科博士課程修了後,東京都医学総合研究所協力研究員,12年UMass Medical School研究員を経て,14年京都大学大学院医学研究科細胞機能制御学特定研究員,18年より現職.
研究テーマと抱負炎症・癌・免疫疾患発症における直鎖状ユビキチン鎖依存的な新たな病態形成機構の解明を目指す.最近は癌内炎症の成因や維持機構に興味を持ち,独自の研究を進めている.
ウェブサイトhttps://www.mcp-kyoto-u.jp/