脳は1000億もの神経細胞から構成されるネットワークであり,このハードワイアこそが感覚,運動,思考のあらゆる源になっている.脳研究,神経科学は今や多岐の分野にわたるが,この神経細胞によるネットワークがどのようにして形成されるかは,最も重要な問題の一つである.
1960年代初めに,Sperryはカエルの視神経が切断後に再生する際,眼球を180度回転させても元の脳部位に正しく到達することを発見し,軸索と標的細胞間の化学物質による親和性が正しい神経回路の形成に必要だとする化学親和仮説(chemoaffinity hypothesis)を提唱した1).この仮説は,その後の分子機構を追求する研究の基礎となった.実際,生化学的手法や細胞生物学的手法を用いた研究によって,その仮想分子の存在や物質的特性は明らかになったが,同定されるまでにはそれなりの年月が必要だった.1980年代に入ると,神経科学にも本格的に分子生物学の波が押し寄せ,1990年代についに軸索成長を誘引する因子や逆に抑制する因子が見いだされるようになった2).引き続き,さまざまな神経系で軸索成長関連因子やそれに対する受容体が次々に発見され,現状は軸索誘導を担う分子機構の基本原理が解明された状況で落ち着いているといってよいだろう.
一方,神経回路を構築するためのもう一つ重要な要因として,ニューロンの電気的活動があげられる.HubelとWieselは,半世紀以上前に視覚入力によって大脳皮質視覚野の回路が変化することを示した3).両眼視が発達した哺乳類では,左右の眼に対応する外側膝状体(網膜からの視覚入力を中継する視床領域)からの線維が視覚野に交互に入る特有の投射パターンが形成される.ところが,幼児期に単眼を遮蔽すると,開眼側の外側膝状体軸索はその投射領域を拡大し,閉眼側のそれは縮小する.しかも,その状態が成体になっても固定化されるのである.同様に,発達段階での視覚体験が皮質ニューロンの方位選択性を担う神経回路形成に影響を与えることも報告されている4).このような大脳皮質視覚野における知見は,感覚による活動が脳内の神経回路を形成する上で重要であることを明確に示している.
しかし,必ずしも感覚由来の神経活動だけが重要なのではない.脳内では自発的な発火活動が発達初期から生じ,この自発発火活動も神経回路形成に決定的な役割を演じている5–7).Sperryの化学親和説の元となった網膜から視蓋の投射においては,その後の分子生物学的なアプローチから,受容体型チロシンキナーゼの一つであるEph受容体とそのリガンドであるEphrinの相互作用が重要な役割を果たしていることが明らかにされているが8),これだけでは精密な投射パターンは形成されない.マウスやラットでは網膜から視蓋への投射は生後約1週間で完成するが,この生後発達の期間に網膜神経節細胞では自発的な発火活動が一定の頻度で生じ,この自発発火パターンが消失すると,特異的な投射形成が阻害される9).すなわち,感覚入力だけでなく自発発火活動も神経回路形成に重要な役割を果たすのである.
このような研究の流れの中にあって,筆者らは特異的・可塑的な神経回路形成の問題に取り組んできた.研究者はそれぞれ,得意な方法論を駆使して研究を進めるが,筆者はin vitroの実験系を駆使してきたのが特徴といえよう.方法論的なブレークスルーは画期的な発見につながるが,研究者人生でそうそう起こることではない.局面局面で,小さくとも工夫した実験系の構築や,全体を通しての研究戦略は重要になる.このことを踏まえて,本稿ではこれまでの長期にわたる取り組みを紹介したい.
当初の研究目的は,哺乳類において脳内の代表的な神経回路である視床から大脳皮質への軸索投射の形成を担う細胞・分子メカニズムを明らかにすることであった.そのために,どのような実験系が適しているかについてまず取り組んだ.そして,in vitroの実験系を開発することにした.現代の神経科学を含む生物学の研究においては,in vivoにおける決定的な証拠を提示することは不可欠であるが,いきなりそれに挑むことに非合理性があることも多々あり,加えてin vivoでの研究自体が困難な場合もある.そういう意味で,組織培養法を用いたin vitro研究は実験条件を単純化するだけでなく,種々の実験操作が容易であることなどから,依然として有用な方法であるといえよう.
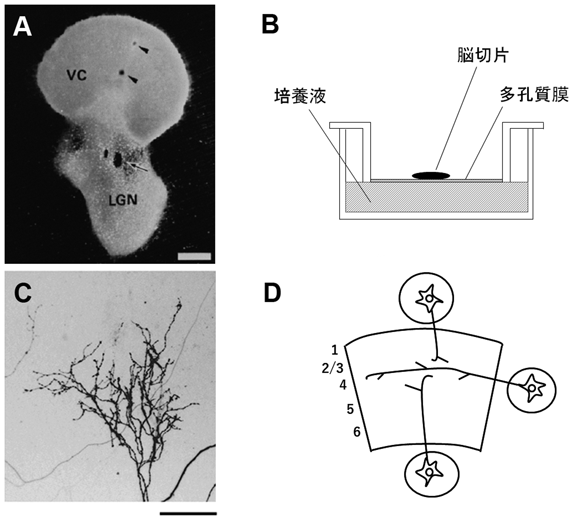
一方で組織培養法は,in vivoの脳内でみられるような構造に依存した特異的な神経回路を構築するには必ずしも適してはいなかった.しかし,1980年代になって脳から単離した海馬組織を用いて細胞構築を維持した状態で培養する方法が開発された10).Gahwilerは海馬組織をカバーグラスに接着させ,それを少量の液とともに回転させながら培養することによって,その層構造や錐体細胞などの形態を維持し,かつ特徴的な神経回路を再構築できることを示した.ただし,この回転培養法も脳切片の接着にコツがあり,言うほど簡単ではなかったため,筆者らはその方法を改良することを試みた.そもそも,回転培養法の肝は,組織片が培養液の供給を受けると同時に,培養室内のガスと接することによってガス成分の供給がより大きくなることにある.1980年代後半,ちょうど抗体染色などを主たる目的として開発された通気性に優れた多孔質膜を利用したプラスチック皿が市販されるようになり,それを用いることにしたのである.すなわち,組織片をその多孔質膜の上に配置し,その膜直下に培養液を満たしておくのである(図1).改良といってもそれだけのことではあったが,思いのほか組織片はその条件下で安定して生存することがわかった.実際,この方法でラット大脳皮質切片と感覚の中継核である視床との共培養を行い(図1),大脳皮質特有の層状構造を維持した状態で,視床–大脳皮質間の層特異的な神経回路を培養皿内で構築させることに成功した11, 12).
ただし,この培養法を確立するには問題点もあった.研究目標は神経回路形成を制御するメカニズムの解明であり,大脳皮質と視床は回路が完成される前に動物から取り出すことが理想であるが,そう簡単に問屋は卸してくれない.時期の制限が大きいのである.齧歯類では出生前から視床軸索は成長し始め皮質に到達するが,その時期大脳皮質ニューロンの産生は完了していないだけでなく,皮質の層構造も形成途上にある.胎生期から大脳皮質切片を切り出して培養してもin vivoのような層構造にはならない.これが今でもこの方法の限界かもしれないが,生後にある程度層構造が完成してからでないと,その層構造をin vitroで再現できない.しかし,生後の日数が経つにつれて,培養後の細胞生存率は低下するため,大脳皮質は生後1~2日のラットから切り出した.一方,視床は生後早期に取り出しても生存することが困難であったため,胎生期15~17日のラットから切り出した.このように時期の異なる二つの組織片を培養することとなったが,視床から成長した軸索は,in vivoでの投射パターンと同様に皮質切片の特定の層に投射することがわかった.
皮質の層構造を見いだすためには,ニッスル染色とブロモデオキシウリジン(BrdU)による標識を用いた.ニッスル染色は,サンプルの固定後に細胞体の形状を可視化する古典的な染色法であるが,現在でも最終的に決定された層構造の同定に用いられている.しかし,ニッスル染色によって層構造を判別するには熟練の眼を要するので,同じ日に最終分裂を完了した細胞を標識できるBrdUを用いることで,より客観的に層構造を同定可能になった.これらの方法により,生後間もない時期のラットから得た大脳皮質切片が培養下において,in vivoと同様な層状構造を形成・維持することがわかった11).
次に,視床軸索を観察するため,当時は緑色蛍光タンパク質を用いる技術は開発されていなかったので,脂溶性の蛍光色素DiIを用いた.視床軸索は皮質細胞の柱状構造に沿って侵入し,その多くは皮質第4層に対応する付近で分岐していた(図1C).実際,哺乳類の大脳皮質感覚野では,個々の視床軸索は第4層で複雑な分岐を形成する.さらに,視床を電気刺激したときに生じる皮質での電場電位(多数の細胞のシナプス電位や活動電位の集合)を解析すると,皮質第4層付近に大きな電場電位が生じていることがわかった11, 12).以上の結果は,視床軸索と皮質細胞間に適切な軸索の成長およびシナプスの形成を導く制御機構が存在することを示している.
3. 培養下の解析による層特異的な神経回路形成メカニズムの研究
1)タイムラプス観察による軸索動態の解析
次なる問題は,皮質内に侵入した軸索がどのようにして層特異的な投射を完成させるかである.そのためのアプローチとして,視床軸索の動態を調べることはきわめて有効である.このころちょうどnetrinやsemaphorinなどのガイダンス分子が同定され始めた時期であったが2),分子機構に迫るためにも,軸索伸長の挙動を調べることはその手がかりを与えてくれるものとして期待した.このためには,個々の軸索の成長・動態を時間軸に沿って調べることが必要であった.視床–大脳共培養において,培養1週間後に蛍光色素DiIの極小結晶を視床組織片に埋め込むことによって一部の視床軸索だけを標識し,生きた状態で共焦点レーザー顕微鏡を用いて観察した.顕微鏡のステージ上には,炭酸ガス培養器内と同様な環境(5%炭酸ガス,温度37°C)を保持するチェンバーを手作りで準備した13).in vitroならではの実験として,視床組織片と大脳皮質切片の位置関係を自在に変えることも可能であった(図1D).まずは,本来の位置関係として,視床組織片を皮質切片の脳室側に配置して軸索の挙動を調べた.脳質側から侵入した視床軸索は深層を一定の速度で伸長するが,第4層付近に到達すると急激に成長速度を低下させた.このことは,軸索成長を停止させるシグナルがあることを示唆している.面白いことに,成長の停止は髄膜側(第1層側)から侵入した場合にもみられたが,層と平行に侵入してきた軸索では起こらなかった(図1D).おそらく視床軸索は標的層と隣接する層との相対的な分子環境の差を識別していると考えられる.
神経回路が形成されるための軸索の挙動でもう一つ重要なのは分岐形成である.一般に中枢神経系では軸索は標的部位で分岐を形成して,多数の細胞とシナプスを介して連結する.タイムラプス観察によって,視床軸索は過たず最初から第4層付近だけで枝分かれを形成することが示された13).さらに,成長停止とは異なって,皮質切片のどの方向から侵入しても,標的層で枝分かれを作り始めることもわかった(図1D).加えて,第4層で停止し損なっても枝分かれ形成が生ずることから,軸索分枝と軸索成長の停止を担う分子機構は互いに独立していると考えられる.
2)in vitroシミュレーションを用いた軸索成長・分岐の解析
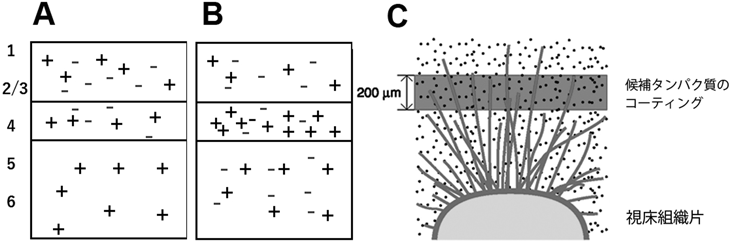
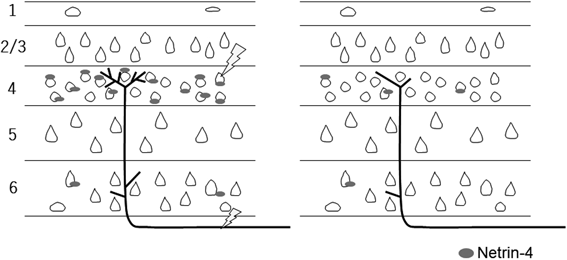
軸索が特定の層で伸長を停止するためには,層ごとの伸長活性に差があるはずである.また,これらの活性は細胞膜表面あるいは細胞外基質に分布する分子に起因する可能性が高い.なぜなら,タイムラプス観察により,軸索の伸長停止が特定の層で急激に起こることがわかったからである.このような細胞表面分子による軸索伸長制御の分子機構を調べるために,皮質切片をホルマリンで短時間固定する手法を採用した14, 15).この手法では,液性タンパク質の関与はなく,細胞表面のタンパク質や糖鎖などの抗原分子がある程度は保持されると考えられる.さらに,固定後にさまざまな酵素処理を加えることによって,特定の分子群の役割を解析することが可能である.実際,生後7日目のラット大脳からホルマリン固定皮質切片を作製し,その側方に未固定の生視床組織片を配置すると,軸索は層と平行に伸長するが,表層(第2/3~4層)での成長は深層に比べてかなり低かった13).ところが,固定切片を培養に先立ってホスファチジルイノシトール特異的ホスホリパーゼC(グリコシルホスファチジルイノシトール結合を介して細胞表面に存在するタンパク分子などをその結合部位で切断する酵素)によって処理すると,深層での成長は変わらずに,表層での成長が顕著に増加した.これらの結果は,グリコシルホスファチジルイノシトール結合型の分子による軸索成長抑制活性が表層に存在することを示唆している.これによる深層と表層との軸索成長活性の差が,視床軸索を第4層で停止させるのに貢献しているかもしれない(図2).
さらに筆者らは,層ごとに異なる分子が存在することを想定して,皮質の層構造と同様なサイズ(100 µm単位)で複数の細胞表面分子をニトロセルロース膜に塗布するシステムを開発し(マイクロプリンティング,図2C),この膜上で視床組織片を培養し,成長する軸索の挙動を調べた16).このプリンティングはオフセット印刷の原理に基づいており,理想科学工業(年賀状作製で人気があった「プリントゴッコ」を製造)の協力によって可能になった.一種のシミュレーションといってよいだろう.この実験と並行して特定の層に発現する遺伝子の探索を行い,皮質第4層に強く発現する遺伝子としてSema7AやKit ligandが候補にあげられていたので,これらの発現ベクターを用いてタンパク質を産生・精製し,それをマイクロプリンティングした.このプリントしたニトロセルロース膜上で視床軸索の成長を観察すると,Sema7AやKit ligandをプリントした領域で軸索成長が止まること,あるいは成長がより促進されるが見いだされ,層ごとの異なるタンパク質の発現パターンによって,軸索の成長がコントロールされていることが示された.
軸索成長の促進あるいは抑制に関する分子機構の研究が世界中で進展していく中,軸索分岐の研究は後れをとっていた.2023年現在でもそれほど理解が進んだとはいいがたいかもしれない17).筆者らは,上述した固定皮質切片を用いて視床皮質投射での軸索分岐を担うメカニズムについても解析を試み18),生きた視床細胞の軸索が,固定切片上でも過たず標的である第4層で分岐を形成するという結果を得た.このことは,皮質の細胞表面分子あるいは細胞外基質分子によって軸索分岐が制御されていることを強く示唆するものである.さらに,固定切片を細胞接着因子(neural cell adhesion molecule:NCAM)に結合する糖鎖,ポリシアル酸(polysialic acid:PSA)を特異的に取り除く酵素で処理して視床軸索を伸長させたところ,第4層以外での枝分かれが対照群に比べて著しく増加したのである.すなわち,NCAMに結合するポリシアル酸が全層に分布し,標的層以外でよけいな枝分かれが形成されるのを阻止していると解釈できる(図2B).ただし,依然,標的層での分枝が支配的であることから,第4層には枝分かれ形成を促進する分子が局在していると考えられる.
4. 遺伝子導入法による軸索標識・神経活動操作を用いた神経活動依存的な軸索分岐メカニズムの研究
1)緑色蛍光タンパク質による軸索標識
最初に述べたように,神経活動に依存した回路形成機構があることは古くから知られていたが,そのメカニズムの解明は今なお中心的な問題である.我々は,層特異的な回路形成機構に加えて,神経活動依存的な問題にも挑戦することにした.神経活動依存性の研究を進展させようとしたのには,軸索ガイダンス機構の研究が世界的に頭打ちになってきたことも一因であった.さて,その最も基本的な問題は,感覚由来,自発発火にかかわらず,発火活動が軸索分岐に対してどのように作用するのかということである.このシンプルな問いに答えるために,再び視床–大脳の共培養系で明らかにしようと考えた.
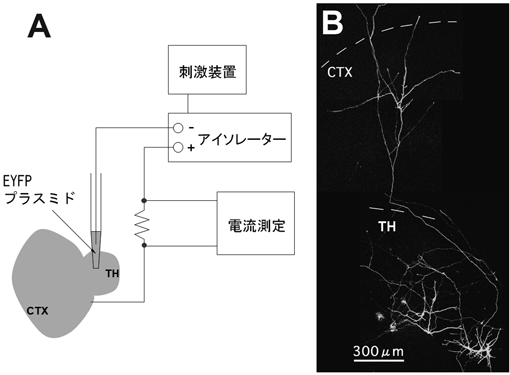
視床軸索の可視化には当初は蛍光色素を使用していたが,必ずしも鮮明に軸索を標識できるわけではなかった.そのような中,緑色蛍光タンパク質(green fluorescent protein:GFP)による細胞標識法が利用できるようになってきた.特に,電気刺激による遺伝子導入法(電気穿孔法)は,分子生物学的手法を敷居が高いと感ずる筆者たちにも身近なものだったのでそれに飛びついたのだが19),当時発生学の実験で用いられた平板金属電極による電気穿孔法は多くの細胞を標識するのに適しているが,少数の細胞に遺伝子導入するのには向いていなかった.本研究で望むべきことは,個々の視床細胞から発する軸索の形態をつぶさに明らかにすることであるにもかかわらずである.この問題を克服するためには,電気穿孔法の改良から始めることになった.大学院生とともに,微小電極を培養脳切片に対して配置することを日々試した.最初は,タングステンの双極電極を用いて,さまざまなパルス波を試した.上述した大型電極では1アンペア近い電流値が必要であるが,双極電極では1万分の1のオーダーで十分であること,高頻度のパルス波を繰り返し与えることが有効であることなどがわかるようになった.ただし,金属電極では組織へのダメージが強いため,ガラス電極を用いることにした.さらに,口径が比較的大きい方が標識されやすいことなどにたどり着くのには試行錯誤の連続であった.その結果,最終的に落ち着いたのが,あらかじめプラスミド溶液を標識したい位置に滴下あるいは注入し,その直後にガラスピペット(プラスミド溶液あるいは塩類溶液を充填)を組織に密着させて,200 Hz, 1秒のパルス波を与える(約0.5マイクロアンペア)という方法だった(図3A).
この電気穿孔法により,培養開始後1~2日に蛍光タンパク質の発現プラスミドを視床細胞に遺伝子導入した20, 21).本実験では,蛍光タンパク質として蛍光強度の強いenhanced yellow fluorescent protein(EYFP)を用いた.生きた状態で観察できるのは蛍光色素と同じであるが,蛍光色素を用いた場合は時間が経つに連れて,背景の蛍光強度が徐々に上昇するのに対して,蛍光タンパク質による標識では一切そのような状況にはならない.蛍光強度も強いため,細い軸索の末端まで観察できるのはもちろんである.その結果,この標識法によって視床軸索が皮質切片に侵入し第4層付近で分岐を形成することを再確認できた(図3B).
軸索標識法だけでなく,本実験では神経活動を操作あるいは記録することが求められる.それを達成するために,視床–大脳皮質切片を多電極培養皿上で作製し,形態学的な変化と神経細胞の発火活動を同時に解析できるようにした21).培養後約1週間から自発発火が生じ始め,培養2週後には,視床,皮質両方とも平均1 Hz以上の発火が観察されるようになった.この自発発火活動の特徴は,視床と皮質で同期的に発生することであった.しかも,自発発火活動が活発になる時期は軸索分岐が形成される時期に一致するので,この自発発火が軸索分岐に作用しているのではないかと考え,薬理学的な手法によって神経活動をブロックし,それによって軸索分岐が変化するかを調べた.実際,Naチャネルの阻害剤であるテトロドトキシン(tetrodotoxin:TTX)を培養1週後に加え,培養2週後に軸索分岐を観察したところ,視床軸索の分岐形成が顕著に抑制されることが判明した.グルタミン酸受容体の拮抗剤によってシナプス伝達を止めてもTTXと同様な効果があったことから,シナプス活動も分岐形成に関与していることが示唆された.
軸索分岐の形成過程で興味深い様相は,分岐が形成される際に新たな枝が出現するだけでなく,いったん形成された枝が消失する,すなわち負の現象も並行して起こっていることである.実際,視床–大脳共培養標本で標識した視床軸索を毎日観察すると,新たな枝が次々と出現する一方で,消失する枝も多数出現する.発達過程では枝の出現や成長が消失や縮退を上回るために全体としては徐々に枝が複雑化するが,枝の出現・成長という正の変化と縮退・消失という負の変化の両方が神経活動によって促進されることも明らかになった21).
2)チャネル分子の遺伝子導入による神経活動操作
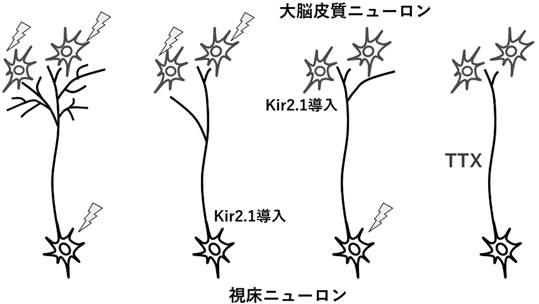
神経活動が軸索分岐を促進するといっても,視床と大脳皮質神経細胞のどちらの神経活動が分岐に作用するのであろうか.神経生理学的な観点から,それがプレシナプス側に原因があるのかポストシナプスによることなのかは重要な課題である.分子機構に迫る上でも詰めなければならない点である.しかし,薬理学的な実験では加えた薬剤が培養細胞全体に作用するため,視床と皮質神経細胞の活動を別々に制御することは事実上不可能である.この仕分けのために,内向き整流性カリウムチャネルKir2.1(現鹿児島大学・田川義晃氏から供与)を含むプラスミドを遺伝子導入法により,視床あるいは皮質神経細胞に導入することにした22, 23).
Kir2.1を導入した細胞では,膜電位が過分極し,自発発火活動がほとんど起こらなくなる.視床神経細胞にEYFPとともにKir2.1を導入し,その細胞から成長する軸索形態を調べると,枝分かれが顕著に抑制された.一方,多数の皮質細胞にKir2.1を導入して皮質切片全体の発火活動を抑制した場合でも,EYFP標識された視床軸索の分岐形成は抑制された.TTX存在下で軸索分岐がほとんど起こらなくなったことを考え合わせると,これらの結果は,軸索の分岐形成には成長する軸索側(シナプス前細胞)ならびに受けて側の細胞(シナプス後細胞)の両方の発火活動が必要であることを示唆している(図4).しかし,これらはいずれも神経活動を抑制することによる軸索動態の変化を観察する実験であることから,証拠としては不十分である.すなわち,神経活動を増加させたときの枝分かれを解析することが求められる.論文としては後になってから発表した証拠であるが,原核生物由来膜電位感受性ナトリウムチャネル(NaChBac)を視床細胞に導入して神経活動を増大させ軸索分岐を調べたところ,予想どおり分岐数は著しく増加したのだ24).このように,軸索分岐数は,神経活動の強度に大まかには比例して増減することが明らかになった.
以上の結果とは正反対であるが,神経活動は軸索分岐を抑制し回路を安定化させるという報告も少なからずある25–27).網膜視蓋投射においては,網膜神経節細胞の軸索分岐がTTX投与によって促進されるのである.この違いは何からくるのか.発達時期の違いによるのかもしれない.調べられた網膜視蓋投射では,軸索分岐が形成され始めた後にTTXが投与されていた.視床–大脳共培養の実験でも,培養後2~3週間を経過して複雑な枝分かれが形成されてからTTX投与を行うと,分岐は減少するのではなく,増加する傾向が認められている(未発表).したがって,軸索分岐が形成され標的細胞と十分なシナプス結合を持った状態と,シナプス結合が少ないあるいは未熟な状況では,神経活動が軸索分岐に及ぼす効果が異なるのかもしれない.
5. 神経活動依存的な軸索分岐の分子機構解明のために
1)標的細胞側の分子機構解明に必要な種々の方法論
では,神経活動依存的な視床軸索の分岐を制御する分子メカニズムはどのようなものであろうか.前述したように,それは視床細胞側と大脳皮質細胞側それぞれに存在すると考えられる.大脳皮質側の分子メカニズムとしては,大脳皮質細胞から軸索分岐を促進する分子が神経活動依存的に発現し,それが視床軸索に作用するとの仮説を掲げた(図5).本研究に先立って発達期大脳皮質に発現する遺伝子を探索した結果から,複数の分泌性因子や膜タンパク質(視床軸索に対して作用しうる)が皮質表層に発現することを見いだしていた28).その中で神経活動によって発現が変化するものを調べたところ,Netrinファミリーに属するNetrin-4が浮かび上がった29).
その神経活動依存性を明らかにするために,netrin-4の遺伝子座にnetrin-4の代わりにβ-galが組み込まれた遺伝子改変動物(北海道大学北田一博氏との共同研究)を用いて解析を行った30).この動物を異なる視覚環境で飼育し,大脳皮質視覚野でβ-galの発現を調べたのである.生後3週間正常な明暗サイクルで飼育したグループ,3週間の暗室飼育(視覚入力の遮断により大脳皮質視覚野の細胞における活動電位の発生は極度に低下すると考えられる)したグループ,そして生後3週間の暗室飼育後に1日明環境に置いたグループの三つの条件で,β-gal陽性細胞の数を調べた.その結果,暗環境ではβ-gal陽性すなわちnetrin-4発現細胞の数は,正常な視覚環境で飼育したものに比べて顕著に減少した.一方,暗環境後1日明環境のグループでは逆に有為に増加した.さらに,培養標本において薬理学的実験によって神経活動を変化させると,TTX投与群ではnetrin-4発現細胞数が著しく減少し,逆にKC1添加の脱分極により有意に増加した.これらin vivoとin vitroの結果は,神経活動が活発化するとNetrin-4の発現が増大することを強く示唆するものである29).
Netrin-4は視床軸索に対してどのように作用するのであろうか.視床–大脳皮質共培養標本にNetrir-4タンパク質を添加し視床軸索の分岐を調べた結果,視床軸索の分岐数はおおよそ倍増することが判明した.さらに,その内在的な作用をnetrin-4欠損動物の視床皮質投射系で解析した.野生型およびNetrin-4欠損動物で,外側膝状体に軸索トレーサー,biotinylated dextran amineを局所注入し,外側膝状体神経細胞の軸索分岐を大脳皮質視覚野で解析したのである.本実験では,トレーサーを脳の特定領域,外側膝状体に正確に微量注入する技術,またその後の標識軸索の解析技術という二つの難題が含まれている.もちろん,それを自前で行う選択肢もあるが,それには相当な期間を要する.筆者らは,鳥取大学の畠義郎らに共同研究を申し出た.このような共同研究で重要なのは,単に技術的な側面だけでなく,共通の問題意識があるかどうかである.畠らは視覚野における可塑性の研究に長年注力し,学問的興味を共有しているグループなのである.実際,快く共同研究を引き受けていただき,実験は鳥取大学で行われた.期待される結果は,欠損動物において第4層での軸索分岐が減少することであったが,残念ながら全体的な軸索分岐の様相には変化はみられなかった.しかし,根気よく顕微鏡をながめていると,欠損動物では末端部の枝分かれが少ない傾向を見いだし,それを定量的に解析すると,欠損動物では末端での分岐数が有意に減少することがわかった29).
次に,皮質細胞が産生するNetrin-4が視床軸索に作用するメカニズムを調べた.netrinファミリー分子の受容体としては,unc5a, unc5b, unc5c, unc5d, dcc, neogeninが候補としてあげられる.これら受容体の視床での発現をin situハイブリダイゼーションによって調べると,unc5b, dcc, neogeninが視覚や体性感覚の視床領域に強く発現することがわかった.これら3種類の受容体を株細胞に発現させ,Netrin-4タンパク質との結合性を調べる実験を行うと,UNC5BがNetrin-4と強く結合することがわかった.最後に,UNC5B受容体が視床軸索分岐に関与する可能性を,視床–大脳皮質共培養標本を用いて調べた.視床細胞にUNC5BのshRNAを導入し,内在的なUNC5Bをノックダウンすると,視床細胞から発する軸索の枝分かれは顕著に減少した.逆に,UNC5Bを過剰発現させた場合には皮質内でより多くの分岐を形成することもわかった29).
以上の結果をまとめると,大脳皮質において神経活動が活発化すると,皮質細胞でNetrin-4の発現が増大し,UNC5Bを発現する視床軸索がそれに反応して軸索分岐を増大させるメカニズムが存在するのである(図5)29).しかし,Netrin-4だけが皮質細胞側で発現を増加させて軸索分岐を促進しているわけではない.神経活動に依存して発現上昇する分子としてよく知られているbrain-derived neurotrophic factor(BDNF)も視床軸索分岐に促進的に作用することを見いだしている31).
2)軸索側の分子機構とシナプス形成と細胞骨格制御
皮質細胞だけでなく視床軸索側にも神経活動依存的なメカニズムが存在することはすでに述べたが,それは一体どのようなものなのか.この問題に取り組むにあたり,軸索分岐におけるプレシナプス形成に着目し26),視床–大脳共培養において軸索成長と同時にプレシナプスを観察する実験を行った24).プレシナプス特異的に発現するsynaptophysinとGFPとの融合タンパク質とDsRedの遺伝子を視床神経細胞に導入すると,培養1週経過したころからsynaptophysin陽性のドット状構造がDsRed陽性の軸索上に分布するようすが観察された.これらsynaptophysin陽性ドットのほとんどは,視床軸索のプレシナプスに分布するグルタミン酸トランスポーターVGlut2やポストシナプスに局在するPSD95と共局在したことから,シナプス部位を反映していることになる.さらに,synaptophysin陽性点と軸索分岐の形成の関連性を調べるために,タイムラプス観察を行ったところ,新たに出現する枝のほとんどはsynaptophysin陽性点から出現することが判明した.すなわち,軸索分岐が形成される際には,まずプレシナプスが形成され,そのプレシナプス部位を起点として新たな突起が現れるのである24).
このプレシナプス部位特異的な軸索分岐様式が視床軸索の神経活動度に依存して変化する可能性を検討した.そのために,視床細胞にsynaptophysin-GFPとDsRedに加えて,神経活動を減少させるためにKir2.1を発現させ,神経活動を上昇させるためにはNaChBacを発現させた.その結果,軸索分岐はKir2.1の導入により著しく減少したが,逆にNaChBacにより有意に増大した(前述).この結果は,視床軸索の分岐がその神経活動によって制御されることを再確認するものであるが,一方プレシナプス形成は神経活動に比例して増加しなかった.Kir2.1導入によってsynaptophysin陽性点の数は有意に減少したが,NaChBac導入ではほとんど変化しなかったのである.実際,タイムラプスで軸索分岐の形成を観察すると,NaChBacを導入した視床軸索では,synaptophysin陽性のプレシナプス部位以外の位置から枝が出現することが頻繁に見いだされた24).このことは,神経活動に依存して軸索分岐を引き起こす分子メカニズムが変化することを示唆している.一つの可能性として,軸索分岐を担う細胞骨格制御因子が通常プレシナプス部位に局在しているが,神経活動が活発になると,その局在性が変化することが考えらえる.以前に,軸索分岐形成に対して,低分子量GTPase, RhoAが神経活動依存的に軸索分岐を促進させることを示しており32),このような細胞骨格制御因子の局在性が原因となっているのかもしれない.この可能性を検証すべく,RhoAの上流であるRhoGEFの軸索分岐に対する役割についても研究を進めたが,分岐自体への促進作用はあったものの,神経活動依存的な特性を見いだすことはできなかった33).
6. 転写調節因子やエピジェネティック因子の解析へ
ここまで,視床皮質投射において,1)軸索分岐が神経活動によって促進されること,2)その分子機構として,皮質細胞においては神経活動に依存してnetrin-4やBdnfの発現が上昇し,その受容体を持つ視床軸索が反応して分岐を増大させること,3)視床軸索側でも神経活動に依存して細胞骨格制御に関わる分子群が作用して分岐を増加させるメカニズムがあることを述べてきた.さらに筆者らは,これらエフェクター分子の発現を調節するメカニズムに興味を持つに至った.
生理学的・生化学的な研究から,神経活動の活発化に伴い,カルシウムイオンの流入が引き金となって,細胞内でのさまざまな分子経路が活性化されることが示されている.カルシウムイオン濃度の増加によって引き起こされる神経活動依存的なメカニズムとして最もよく知られた系は,転写調節因子cAMP-responsive element binding protein(CREB)によるものであり,ゲノム上のcAMP-responsive element(CRE)配列に結合することにより下流の遺伝子発現が調節される34, 35).CREBは神経細胞においては電位依存性カルシウムチャネルやNMDAチャネルを通るカルシウムイオンによって活性化され,さまざまな可塑的現象に関与している.転写調節因子myocyte-specific enhancer factor(MEF)もカルシウムイオン依存性のシグナルを媒介する34).ただし,MEFは核への出入りではなく,ヒストンによるDNAの修飾作用により制御されている.これらの知見を土台にして,新たな視点を導入することによって研究に取り組むことにした.
1)HDAC9(histone deacetylase 9)に着目したin vitro解析
2002年,ちょうど筆者が独立した研究室を持つタイミングで,DNA修復機構と神経発生との関わりを研究していた菅生紀之氏(当時横浜市立大学理学部)が研究室に合流することになった.神経活動依存的な分子機構に焦点を当て研究し始めたころであったので,転写調節の観点からの研究方向は非常に重要であるとの考えから,発達期大脳皮質の細胞におけるHDACの役割について調べることから始めた.特に,クラス2 HDACはさまざまな転写因子と結合し,核と細胞質間をシャトリングすることで細胞種特異的に遺伝子発現を制御することが知られている36, 37).神経系では,クラス2 HDACの核・細胞質間の移動は発火活動によって引き起こされ,細胞の生存に関与することがすでに示されていたが,発生過程における神経細胞の分化や結合変化における役割は不明であった.
筆者らは,クラス2 HDACの一つであるマウスHDAC9(別名MEF2-interacting transcriptional repressor)による神経活動依存的な制御機構を,マウス大脳皮質神経細胞の分散培養系で調べた38).分散培養下でもスライス培養と同じく,培養1週間ぐらいまでは自発発火活動は低いが,培養2週間が経過すると,自発発火活動は顕著に増大する.培養開始時にHDAC9-EGFP(enhanced GFP)プラスミドを導入すると,培養初期にはHDAC9-EGFPが核に局在しているが,培養2週間後にはほとんどが細胞質へ移動していることが観察された.同様な核・細胞質間移動は,in vivoにおいても観察された.この移動を阻止すると皮質細胞の神経活動依存的な遺伝子発現調節に異常を来し,細胞分化にも影響が出ることが予測される.その可能性を検証するために,核–細胞質間の移動ができないHDAC9変異体を導入し,そのドミナントネガティブな作用を使って調べてみることにした.すると,HDAC9変異体を導入した細胞では,神経活動依存的遺伝子の代表選手であるc-fosの発現が顕著に抑制され,同時に樹状突起の成長が減少した.逆に,内在性のHDAC9をshRNA法によってノックダウンすると樹状突起の成長が促進された.これらのことから,発達期の大脳皮質神経細胞において,HDAC9の核細胞質移動を伴うクロマチンリモデリングが,活動依存的な遺伝子発現と樹状突起成長を制御していることが示唆された38).
そもそもの問題に立ち返って,HDAC9の神経活動依存的な役割が,視床軸索の分岐形成にも当てはまるかを調べた39).視床–大脳皮質の共培養系で,HDAC9-EGFPを視床細胞に発現させると,皮質細胞と同様に培養中に神経活動の増大に伴ってEGFPのシグナルは核から細胞質へ移動した.そしてこの移動ができないHDAC9変異体を発現させると,培養中の視床軸索の分岐が著しく減少した.また,核細胞質移動ができないかつMEF2との相互作用ができないHDAC9変異体を発現させると,軸索分岐が回復することが確認された.加えて,in vivoにおいても変異型HDAC9(前者の変異体)の遺伝子導入により,視床軸索の分岐が顕著に減少することも判明した.ここで簡単にin vivoでもわかったと記述したが,視床への遺伝子導入は世界でもそれをできる人がどれだけいるかというレベルの超越した技術であるため,その先駆者でもある下郡智美氏(理化学研究所脳科学センター)の協力のもと得られた実験成果である40).以上,HDAC9の核細胞質移動は,MEF2による転写調節や下流のシグナル伝達経路に影響を与え,活動依存的な視床軸索分岐に重要な役割を果たすことが示唆された.
2)電気刺激による神経活動依存的な遺伝子発現の解析
神経活動依存的な転写調節を担う分子機構は,基本的枠組みだけでなく,分子経路やクロマチン構造制御の詳細が明らかにされつつある.しかし,これらの研究のほとんどは,いやほぼすべてといっても過言ではないが,生化学的・分子生物学的なアプローチによるものであり,神経科学・神経生物学的な観点に欠けている.具体的にいうと,それらの研究では神経活動を操作するために,培養細胞に対して外液のKCl濃度を増大させる手法が用いられてきた.しかし,神経生理学を少しでも学んだならば,これがいかに生理学的な刺激とはほど遠いかはほぼ自明である.にもかかわらず,実験的な簡便さもあり(といいながら筆者らも使っていることがある),いまだにKCl刺激は健在である.しかし,脳は外界からさまざまな入力刺激を受け,それは神経細胞のシナプス電位や活動電位という電気信号に変換される.したがって,生理学的な観点からいうと,実際の電気的活動に沿った実験を組み立てることが欠かせない.筆者らはこの切り口を一つの突破口にしたいと考えている.
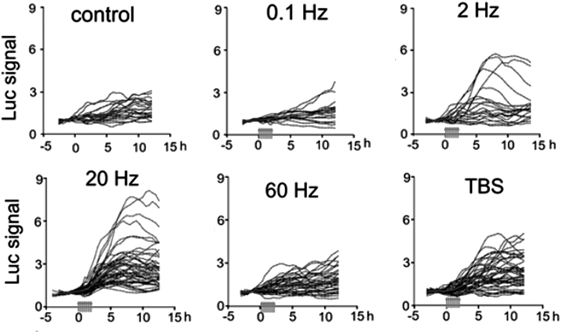
そのための第一弾として,神経活動依存的な回路形成に関与するBDNFに着目し,このプロモーター活性を培養大脳皮質切片の個々の細胞においてライブイメージングし,その標本にさまざまなパターンの刺激を与えるという実験を計画した41).まず,Bdnfのプロモーター領域(エクソン4プロモーターの約300 bpの領域でCREBの結合領域であるCREサイトも含まれている)をルシフェラーゼ発現プラスミドに組み入れたコンストラクトpGL3-Bdnfpを作製し42),ラットあるいはマウス大脳皮質感覚野の脳切片培養に電気穿孔法を用いて散在的に遺伝子導入した.培養1~2週で刺激電極を装着した培養皿に移し(学生の手作り),顕微鏡下でEMCCDカメラによって,個々の皮質細胞の発光を捉えた.遺伝子導入された細胞の半数以上は刺激前においても弱い発光を示すので,それがベースラインとなる.この条件下で,装着された電極を通して,脳波の周波数帯を参照し0.1~60 Hzの定頻度刺激やバースト刺激を与えて,発光強度の時間的変化を測定した.その結果,Bdnfプロモーター活性の増加レベルやタイムコースは刺激パターンによって異なり,増加率は細胞ごとに大きく異なったが,20 Hzやθバースト刺激(theta burst stimulation:TBS)がプロモーター活性の上昇に有効に働くことがわかった(図6).この結果から,個々の皮質ニューロンにおけるBdnf発現量や時間的特性が神経活動パターンによって調節されることが示された41).
3)転写調節因子の1分子イメージング法による時空間的動態の解析
生理学的観点だけでなく,転写調節因子や関連分子の核内での時空間的な動態の解明も重要と考えられるが,その知見も欠けている.転写調節機構に関する論文のほとんどは,チップによる遺伝子座の同定や遺伝子発現データ,それにノックアウトマウスの行動解析が入って,一段落となっているものがほとんどである.もちろん,重要な研究の方向性ではあるが,同じようなやり方で迫っても,研究としては面白みに欠けるだけでなく,新規参入ではそれらを超えるような発見は望みがたい.これらの状況を踏まえて,筆者らは核内の時空間的な分子動態に焦点を絞り,1分子イメージング法を導入することによって問題に取り組み始めた.もう一つ,このような異分野の共同研究が研究科内・学内で推奨されたことが推進力にはなったこともつけ加えたい.実際,1分子イメージングの先駆者である柳田敏雄氏の研究室の方々からの協力によって研究を始めることができた43).
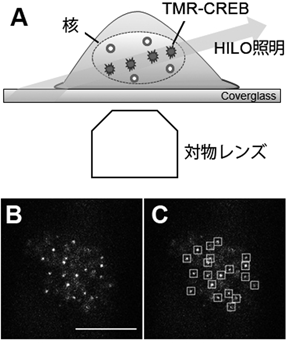
目指すところは,転写因子とその標的DNA部位との時空間的な相互作用に対する神経活動の役割を明らかにすることである.具体的には,1分子イメージング法によって,転写調節因子CREBのDNA結合・解離の神経活動依存性をマウス大脳皮質神経細胞で調べることにした44, 45).実験としては,タグ付けされたCREBを分散培養した大脳皮質細胞に低レベルで発現させ,その1分子動態をHILO(highly inclined laminated sheet optics)照明下で経時的に観察した(図7).2分間の観察中に一つの核内で数千から1万個のCREBスポット(一つ一つのスポットが1分子を表す)が計測され,大多数のCREBスポットは1秒以内で消失するが,5%程度のCREBスポットは数秒間同じ場所に滞在した(図7).それに対して,DNA(CRE配列)に結合できない変異型CREBで同じ実験を行うと,長期間滞在するスポットの数は著しく減少した.このことは,長く滞在するCREBスポットはゲノム上のCRE配列に結合していることを示唆している.ここからが肝心の神経活動によってCREB動態がどのように変化するかである.神経活動上昇によって長時間の滞在を示すCREBスポットの数が増えることを期待したのである.刺激するためには,KCl刺激だけでなくチャネルロドプシン-2を用いた光遺伝学的手法も用いた.しかし,結果は長時間滞在するCREBスポットの数はほとんど変化しなかったのである.何か間違っていないかぐらいの勢いで調べ直しても,その結果は変わらずがっかりしたが,気を取り直して,空間分布を観察することにした.すると,神経活動の上昇後に長期間滞在(1秒以上)するCREBスポットが繰り返し出現する部位が出現したのである.刺激前ではそのようなスポットはせいぜい核に1か所であったのに対して,刺激後には複数の箇所でCREBスポットが頻回出現するようになった.これらの結果は,神経細胞の活動がゲノムの特定の部位でCREBが結合する頻度を増加させることによって,CREB依存的な転写を促進することを示唆している.現在ヒトES細胞由来の大脳皮質ニューロンを作製し,その系においてCREBの1分子の動態だけでなくエピジェネティック因子の動態を解析している.本結果がさらに生化学だけではみることができなかった点を明らかにできることを期待している.
本稿では,筆者の約40年にわたる研究の主たる流れをその戦略・方法論の開発をフィーチャーして紹介してきたが,他にもユニークなアプローチを心がけてきたので,最後に少し列挙したい.大脳皮質への入力線維の特異的結合だけでなく出力線維が標的領域に結合するための分子機構にT-cadherinやsemaphorinシグナルが貢献していること46, 47),視床軸索から放出されるタンパク質VGFやNeuritinが標的である皮質第4層細胞の生存や成長を促進することも重要な発見であった48, 49).加えて,恩師塚原仲晃先生が無念にもやり残された,脳損傷に伴う神経発芽(neural sprouting)の分子メカニズムの一端を明らかにできたことも誇らしく思っている50).共同研究として実ったものもあればそうでないものもあったが,本文中には登場しなかった国内外の研究者にも有形無形の協力を得たことも研究を進める上で重要であった.最後に,紹介した研究の一部はスタッフと博士研究員が主体となったものであるが,大部分は研究室の学生の成果であったことも強調したい.筆者は研究者として決して大物ではないので,究極の研究者を目指す人たちにとって必ずしも本稿が有用ではないかもしれないが,研究人生を楽しんで過ごすことの一助になれば幸いである.
引用文献References
1) Sperry, R.W. (1963) Chemoaffinity in the orderly growth of nerve fiber patterns and connections. Proc. Natl. Acad. Sci. USA, 50, 703–710.
2) Tessier-Lavigne, M. & Goodman, C.S. (1996) The molecular biology of axon guidance. Science, 274, 1123–1133.
3) Hubel, D.H. & Wiesel, T.N. (1970) The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J. Physiol., 206, 419–436.
4) Blakemore, C. & Cooper, G.F. (1970) Development of the brain depends on the visual environment. Nature, 228, 477–478.
5) Pumo, G.M., Kitazawa, T., & Rijli, F.M. (2022) Epigenetic and transcriptional regulation of spontaneous and sensory activity dependent programs during neuronal circuit development. Front. Neural Circuits, 16, 911023.
6) Yamamoto, N. & Lopez-Bendito, G. (2012) Shaping brain connections through spontaneous neural activity. Eur. J. Neurosci., 35, 1595–1604.
7) Nakazawa, S. & Iwasato, T. (2021) Spatial organization and transitions of spontaneous neuronal activities in the developing sensory cortex. Dev. Growth Differ., 63, 323–339.
8) Feldheim, D.A., Kim, Y.I., Bergemann, A.D., Frisén, J., Barbacid, M., & Flanagan, J.G. (2000) Genetic analysis of ephrin-A2 and ephrin-A5 shows their requirement in multiple aspects of retinocollicular mapping. Neuron, 25, 563–574.
9) McLaughlin, T., Torborg, C.L., Feller, M.B., & O’Leary, D.D. (2003) Retinotopic map refinement requires spontaneous retinal waves during a brief critical period of development. Neuron, 40, 1147–1160.
10) Gahwiler, B.H. (1981) Organotypic monolayer cultures of nervous tissue. J. Neurosci. Methods, 4, 329–342.
11) Yamamoto, N., Yamada, K., Kurotani, T., & Toyama, K. (1992) Laminar specificity of extrinsic cortical connections studied in coculture preparations. Neuron, 9, 217–228.
12) Yamamoto, N., Kurotani, T., & Toyama, K. (1989) Neural connections between the lateral geniculate nucleus and visual cortex in vitro. Science, 245, 192–194.
13) Yamamoto, N., Higashi, S., & Toyama, K. (1997) Stop and branch behaviors of geniculocortical axons: A time-lapse study in organotypic cocultures. J. Neurosci., 17, 3653–3663.
14) Yamamoto, N., Matsuyama, Y., Harada, A., Inui, K., Murakami, F., & Hanamura, K. (2000) Characterization of factors regulating lamina-specific growth of thalamocortical axons. J. Neurobiol., 42, 56–68.
15) Yamagata, M. & Sanes, J.R. (1995) Lamina-specific cues guide outgrowth and arborization of retinal axons in the optic tectum. Development, 121, 189–200.
16) Maruyama, T., Matsuura, M., Suzuki, K., & Yamamoto, N. (2007) Cooperative activity of multiple upper layer proteins for thalamocortical axon growth. Dev. Neurobiol., 68, 317–331.
17) Hoersting, A.K. & Schmucker, D. (2021) Axonal branch patterning and neuronal shape diversity: Roles in developmental circuit assembly: Axonal branch patterning and neuronal shape diversity in developmental circuit assembly. Curr. Opin. Neurobiol., 66, 158–165.
18) Yamamoto, N., Inui, K., Matsuyama, Y., Harada, A., Hanamura, K., Murakami, F., Ruthazer, E.S., Rutishauser, U., & Seki, T. (2000) Inhibitory mechanism by polysialic acid for lamina-specific branch formation of thalamocortical axons. J. Neurosci., 20, 9145–9151.
19) Nakamura, H. & Funahashi, J. (2013) Electroporation: Past, present and future. Dev. Growth Differ., 55, 15–19.
20) Uesaka, N., Hirai, S., Maruyama, T., Ruthazer, E.S., & Yamamoto, N. (2005) Activity dependence of cortical axon branch formation: A morphological and electrophysiological study using organotypic slice cultures. J. Neurosci., 25, 1–9.
21) Uesaka, N., Hayano, Y., Yamada, A., & Yamamoto, N. (2007) Interplay between laminar specificity and activity-dependent mechanisms of thalamocortical axon branching. J. Neurosci., 27, 5215–5223.
22) Yamada, A., Uesaka, N., Hayano, Y., Tabata, T., Kano, M., & Yamamoto, N. (2010) Role of pre- and postsynaptic activity in thalamocortical axon branching. Proc. Natl. Acad. Sci. USA, 107, 7562–7567.
23) Mizuno, H., Hirano, T., & Tagawa, Y. (2007) Evidence for activity-dependent cortical wiring: Formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J. Neurosci., 27, 6760–6770.
24) Matsumoto, N., Hoshiko, M., Sugo, N., Fukazawa, Y., & Yamamoto, N. (2016) Synapse-dependent and independent mechanisms of thalamocortical axon branching are regulated by neuronal activity. Dev. Neurobiol., 76, 323–336.
25) Constantine-Paton, M. & Cline, H.T. (1998) LTP and activity-dependent synaptogenesis: The more alike they are, the more different they become. Curr. Opin. Neurobiol., 8, 139–148.
26) Ruthazer, E.S., Akerman, C.J., & Cline, H.T. (2003) Control of axon branch dynamics by correlated activity in vivo. Science, 301, 66–70.
27) Cohen-Cory, S. (1999) BDNF modulates, but does not mediate, activity-dependent branching and remodeling of optic axon arbors in vivo. J. Neurosci., 19, 9996–10003.
28) Zhong, Y., Takemoto, M., Fukuda, T., Hattori, Y., Murakami, F., Nakajima, D., Nakayama, M., & Yamamoto, N. (2004) Identification of the genes that are expressed in the upper layers of the neocortex. Cereb. Cortex, 14, 1144–1152.
29) Hayano, Y., Sasaki, K., Ohmura, N., Takemoto, M., Maeda, Y., Yamashita, T., Hata, Y., Kitada, K., & Yamamoto, N. (2014) Netrin-4 regulates thalamocortical axon branching in an activity-dependent fashion. Proc. Natl. Acad. Sci. USA, 111, 15226–15231.
30) Kitada, K., Ishishita, S., Tosaka, K., Takahashi, R., Ueda, M., Keng, V.W., Horie, K., & Takeda, J. (2007) Transposon-tagged mutagenesis in the rat. Nat. Methods, 4, 131–133.
31) Granseth, B., Fukushima, Y., Sugo, N., Lagnado, L., & Yamamoto, N. (2013) Regulation of thalamocortical axon branching by BDNF and synaptic vesicle cycling. Front. Neural Circuits, 7, 202.
32) Ohnami, S., Endo, M., Hirai, S., Uesaka, N., Hatanaka, Y., Yamashita, T., & Yamamoto, N. (2008) Role of RhoA in activity-dependent cortical axon branching. J. Neurosci., 28, 9117–9121.
33) Sasaki, K., Arimoto, K., Kankawa, K., Terada, C., Yamamori, T., Watakabe, A., & Yamamoto, N. (2020) Rho guanine nucleotide exchange factors regulate horizontal axon branching of cortical upper layer neurons. Cereb. Cortex, 30, 2506–2518.
34) Flavell, S.W. & Greenberg, M.E. (2008) Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci., 31, 563–590.
35) Kornhauser, J.M., Cowan, C.W., Shaywitz, A.J., Dolmetsch, R.E., Griffith, E.C., Hu, L.S., Haddad, C., Xia, Z., & Greenberg, M.E. (2002) CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron, 34, 221–233.
36) Yang, X.J. & Gregoire, S. (2005) Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell. Biol., 25, 2873–2884.
37) West, A.E., Griffith, E.C., & Greenberg, M.E. (2002) Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci., 3, 921–931.
38) Sugo, N., Oshiro, H., Takemura, M., Kobayashi, T., Kohno, Y., Uesaka, N., Song, W.J., & Yamamoto, N. (2010) Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur. J. Neurosci., 31, 1521–1532.
39) Alchini, R., Sato, H., Matsumoto, N., Shimogori, T., Sugo, N., & Yamamoto, N. (2017) Nucleocytoplasmic shuttling of histone deacetylase 9 controls activity-dependent thalamocortical axon branching. Sci. Rep., 7, 6024.
40) Fukuchi-Shimogori, T. & Grove, E.A. (2001) Neocortex patterning by the secreted signaling molecule FGF8. Science, 294, 1071–1074.
41) Miyasaka, Y. & Yamamoto, N. (2021) Neuronal activity patterns regulate brain-derived neurotrophic factor expression in cortical cells via neuronal circuits. Front. Neurosci., 15, 699583.
42) Aid, T., Kazantseva, A., Piirsoo, M., Palm, K., & Timmusk, T. (2007) Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res., 85, 525–535.
43) Sako, Y. & Yanagida, T. (2003) Single-molecule visualization in cell biology. Nat. Rev. Mol. Cell Biol., (Suppl), SS1–SS5.
44) Sugo, N., Morimatsu, M., Arai, Y., Kousoku, Y., Ohkuni, A., Nomura, T., Yanagida, T., & Yamamoto, N. (2015) Single-molecule imaging reveals dynamics of CREB transcription factor bound to its target sequence. Sci. Rep., 5, 10662.
45) Kitagawa, H., Sugo, N., Morimatsu, M., Arai, Y., Yanagida, T., & Yamamoto, N. (2017) Activity-dependent dynamics of the transcription factor of cAMP-response element binding protein in cortical neurons revealed by single-molecule imaging. J. Neurosci., 37, 1–10.
46) Zhao, H., Maruyama, T., Hattori, Y., Sugo, N., Takamatsu, H., Kumanogoh, A., Shirasaki, R., & Yamamoto, N. (2011) A molecular mechanism that regulates medially oriented axonal growth of upper layer neurons in the developing neocortex. J. Comp. Neurol., 519, 834–848.
47) Hayano, Y., Zhao, H., Kobayashi, H., Takeuchi, K., Norioka, S., & Yamamoto, N. (2014) The role of T-cadherin in axonal pathway formation in neocortical circuits. Development, 141, 4784–4793.
48) Sato, H., Fukutani, Y., Yamamoto, Y., Tatara, E., Takemoto, M., Shimamura, K., & Yamamoto, N. (2012) Thalamus-derived molecules promote survival and dendritic growth of developing cortical neurons. J. Neurosci., 32, 15388–15402.
49) Sato, H., Hatakeyama, J., Iwasato, T., Araki, K., Yamamoto, N., & Shimamura, K. (2022) Thalamocortical axons control the cytoarchitecture of neocortical layers by area-specific supply of VGF. eLife, 11, e67549.
50) Chang, L., Masada, M., Kojima, M., & Yamamoto, N. (2022) Involvement of denervated midbrain-derived factors in the formation of ectopic cortico-mesencephalic projection after hemispherectomy. J. Neurosci., 42, 749–761.
著者紹介Author Profile
 山本 亘彦(やまもと のぶひこ)
山本 亘彦(やまもと のぶひこ)深圳湾実験室 客員教授.工学博士.
略歴957年京都市に生れる.80年3月大阪大学基礎工学部卒業.86年博士号を取得,京都府立医科大学助手・講師,2002年より大阪大学生命機能研究科教授.22年より中国深圳湾実験室客員教授.
研究テーマと抱負現在のテーマは,神経活動依存的な遺伝子発現の調節機構.主として神経回路形成・再編の細胞分子機構を研究してきたので,単に遺伝子発現調節だけでなく細胞レベルの機能に結びつけたい.ヒト特有のメカニズムに迫りたい.
ウェブサイトhttps://www.szbl.ac.cn/en/scientificresearch/researchteam/3314.html
趣味サッカー,お笑い.