1)遺伝子発現の全体像
遺伝子発現は転写,翻訳,さらに(m)RNAやタンパク質の分解といった複数のステップで制御されている.この一連のステップにおいて,mRNAやタンパク質が合成される速度や,それらの存在量と寿命の全体像はどのようなものであろうか.実際にこれらの情報をゲノムワイドかつ定量的に計測する研究が2010年ごろから盛んに行われてきた.技術的な側面からも,超並列DNAシーケンス技術[もしくは次世代シーケンス(next-generation sequencing:NGS)]に加え,質量分析,数学の融合により遺伝子発現制御の包括的な視点からの理解が深まりつつある.マウス線維芽細胞NIH3T3では単位時間・細胞あたりに合成されるmRNA量の中央値は,一遺伝子あたり2コピー/時間で存在量は17コピー/細胞,半減期は9時間であることがわかってきた1).(筆者は単一細胞に特定のmRNAはたった数(十)コピーしか存在ないことが直感的に信じられなかったが,これは蛍光in situハイブリダイゼーションによるmRNAの絶対定量結果2, 3)とも一致する値である.)それに対して,タンパク質では合成量と存在量はそれぞれ中央値で1000コピー/mRNA/時間と50,000コピー/細胞もあり,半減期は46時間と長い.したがって定常状態では,転写過程に加え,プロテオーム量増減に対する翻訳過程の寄与はきわめて大きいものと考えられる1).たとえば,ヒト単球由来細胞株THP-1が単球からマクロファージ様細胞に分化する過程では,タンパク質の相対的な合成速度の変化がプロテオームの質と量を規定する主要な要因である4).このように翻訳制御は転写を伴わずプロテオーム量を調節できるため,摂動に迅速に応答するための重要なステップの一つでもあると考えられている.
2)翻訳速度
哺乳類細胞におけるリボソームによるアミノ酸の伸長速度は,リアルタイム一分子イメージングの結果5–8)から3~10アミノ酸/秒,リボソームプロファイリングの結果9)から6アミノ酸/秒と算出されている.ヒトプロテオームのタンパク質長の中央値が約300アミノ酸なので,伸長速度を仮に10アミノ酸/秒とすると,時間あたりの合成量はざっくりと120コピー/mRNA/時間となる.これまた上述1)の質量分析から得られた合成量(だいたいのタンパク質が100~1000コピー/mRNA/時間)1)と近い値になる.つまり,三つの異なる手法,質量分析,リボソームプロファイリング,イメージングを用いても,ほぼ同等の翻訳速度値が推測されている.
細胞にとって目的のタンパク質を増やす手っ取り早い方法は,(転写を介さず)既存のmRNAを翻訳する,または定常状態時よりも翻訳速度を上げることである4, 10).実際に,特定のmRNAに対しては定常状態では翻訳は抑制されており,必要に応じて翻訳が誘導される1, 11).もう一つの有効な手段は,タンパク質の分解を抑制することである.たとえば,HIF1α(hypoxia-inducible factor 1α)12)は常にユビキチン–プロテオームにより分解されているが,低酸素状態になると安定化し,その結果,細胞内のHIF1α量が増加することが知られている.
本稿では遺伝子発現における「翻訳」過程に焦点を当て,翻訳をタンパク質レベルで捉えるための最先端の質量分析技術を紹介するとともに,それらの包括的解析から得られた知見について概説する.
まず,mRNA量とタンパク質量の相関について簡単にふれておきたい.オミクス関連技術の発展に伴い,同一試料(細胞株から組織,動物まで)からのトランスクリプトーム・プロテオームの同時解析が可能になり,mRNA量とタンパク質量の相関が至るところで議論されてきた13–17).ここでは混乱を避けるために「遺伝子間相関」と「遺伝子内相関」に分けて説明する.
1)遺伝子間相関
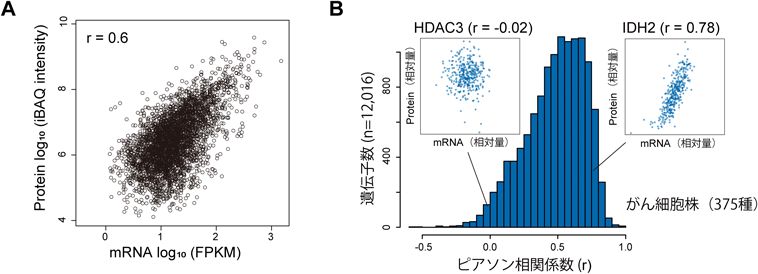
ある特定の条件下で,さまざまな遺伝子由来のmRNAの絶対量と対応するタンパク質の絶対量の相関を遺伝子間相関と定義する(図1A).横軸がmRNA量[例:NGSのリードカウントから得られるFPKM(per kilobase of transcript per million mapped reads)],縦軸がタンパク質量(例:質量分析から得られる正規化シグナル強度)で,各点が遺伝子を示す散布図がそれに相当する.この遺伝子間相関に関しては,さまざまな生物種・細胞・組織において“そこそこ”相関する(ピアソン相関係数で0.4~0.8の範囲)ことがわかってきた13–17).各測定法のバイアス(固有のPCR増幅率やペプチドのイオン化効率など)や実験誤差を加味すると,実際はもう少し高い相関になるはずである.哺乳類細胞におけるタンパク質の濃度差のダイナミックレンジは107~108程度1)であることを考慮すると,散布図の各点(遺伝子)は広範囲に広がるためこれも正の相関関係をもたらす一つの要因ともなる.これらの結果は特定の条件下では,任意の遺伝子に関してmRNA量から対応するタンパク質量を“そこそこ”推測できることを意味している(同時に,mRNA量からその量を予測できないタンパク質も“そこそこ”ある).技術的な側面と生物学的な側面からさらなる検証が必要であり,技術的なバイアスによって生じる誤差17)と転写後制御・翻訳(後)制御によるタンパク質量の調節機構16, 18)を明確に区別しながら議論する必要がある(次の遺伝子内相関も同様である).
2)遺伝子内相関
ある特定の遺伝子に対して,異なるさまざまな条件下(さまざまな刺激や細胞・組織種など)でのmRNA量とタンパク質量の相関を遺伝子内相関と定義する.異なる条件間での,ある遺伝子のmRNAレベルの変化は,対応するタンパク質レベルの変化をどの程度説明できるであろうか? これに関しては遺伝子によってケースバイケースである.図1Bに375種のがん細胞株間19)での遺伝子内相関係数の分布を示す.たとえば,IDH2(isocitrate dehydrogenase)は正に相関し(r=0.78),HDAC3(histone deacetylase 3)は相関がみられない(r=−0.02)(この図では,各点は遺伝子ではなく細胞株を示す).遺伝子内相関の場合,特定の遺伝子に着目するため,mRNA量とタンパク質量の濃度差のスケールが小さい場合も多く,分布が狭い範囲に偏ることも遺伝子間相関と比較して弱い相関を示すことが多い原因となる.
タンパク質量を規定する主要なパラメーターはmRNA量,翻訳速度,タンパク質の半減期である1, 4, 20).仮に翻訳速度とタンパク質の半減期が遺伝子固有の値であれば,原理的にはmRNA量からタンパク質量を予測することができる.しかし,遺伝子内相関をみる限りこれは成立しない.つまりコンテキストに依存した転写後・翻訳(後)制御が背面下に存在しうることを示しており,「何がmRNA-タンパク質量を制御しているか?」という本質的な問いに対してゲノムワイドな視点から迫る研究が今後必要である.
3. 新生タンパク質・ポリペプチド鎖のプロテオームワイドな定量
質量分析技術とケミカルツールの進展に伴い,タンパク質合成をプロテオームワイドに測定するさまざまな手法が開発されてきた13, 21).古典的には35S-メチオニンなどの放射性同位体で新生タンパク質を標識し定量していたが,安全面や発現量の高い(SDS-PAGEでバンドとして確認できる)特定のタンパク質の合成量しか定量できないといった問題がある.一方,安定同位体標識と質量分析を組み合わせることで数千~1万種もの個々のタンパク質の合成を捉えることが可能になってきた.なお本稿ではリボソームで合成過程にあるタンパク質を「新生ポリペプチド鎖」,新生ポリペプチド鎖も含む合成された直後の比較的若いタンパク質のプールを「新生タンパク質」と定義する.
1)安定同位体アミノ酸を用いたpSILAC法
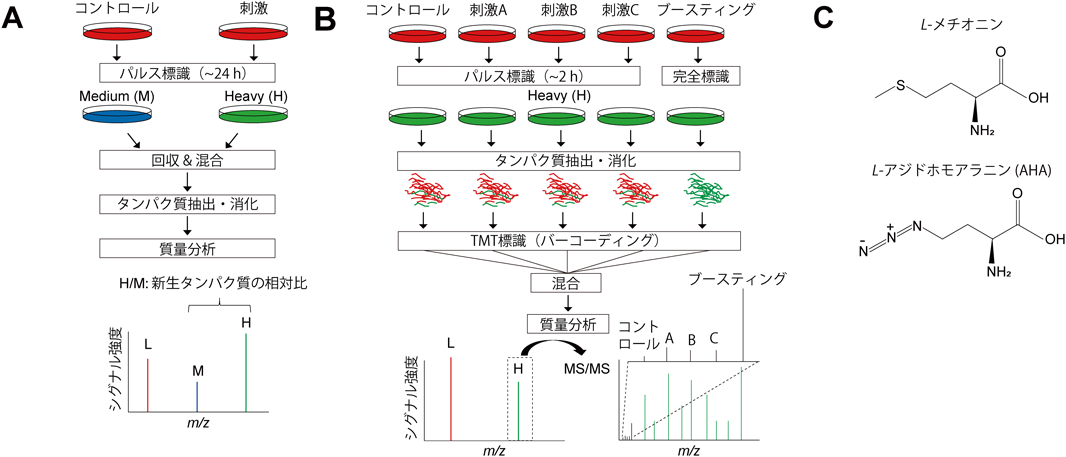
2008年に報告されたpulsed stable isotope labeling by amino acids in cell culture法(pSILAC法)22)では,コントロール群と実験群の二群の細胞を,それぞれ質量が異なる安定同位体アミノ酸[Medium(M)アミノ酸:13C6-アルギニンとD4-リシンのペアと,Heavy(H)アミノ酸:13C6,15N4-アルギニンと13C6,15N2-リシンのペアがよく利用される]を含む培地で24時間程度培養する(図2A).アルギニンとリシンは必須アミノ酸であるため,新しく合成されたタンパク質はすべて安定同位体アミノ酸で標識される.その後,二つの細胞群を混合し,タンパク質抽出,トリプシンによるペプチドへの消化を行い,質量分析を実施する.質量分析計で検出された“H”または“M”標識されたペプチドの相対比(H/M)は,二つの細胞群で合成されたタンパク質の相対比を示し,数千もの個々のタンパク質の合成量を比較できる.pSILAC法が初めて導入された研究22, 23)では,miRNA処理,未処理の細胞を解析し,miRNAによるタンパク質合成抑制を見事に定量的に示しており,単一種のmiRNAは数百のタンパク質を標的とするが,その抑制効果は比較的穏やか(変化量は2倍以下)であることが実験的に初めて示された.
2)pSILAC法とミトコンドリア翻訳
細胞質リボソームとミトコンドリアリボソームはそれぞれ核ゲノムとミトコンドリアゲノムにコードされているタンパク質を合成し,その結果,真核生物のミトコンドリアプロテオームが形成される.最近,筆者らはミトコンドリアの粗精製とpSILAC法を組み合わせることで,ミトコンドリアリボソームで合成される13種の膜タンパク質(以下“MTタンパク質”と呼称)を定量する手法を確立した24).OXPHOS(oxidative phosphorylation)複合体は,ミトコンドリア由来のMTタンパク質と核にコードされているタンパク質で構成されるユニークな複合体である.ミトコンドリアリボソームの翻訳を阻害するクロラムフェニコールでHEK293T細胞を処理すると,MTタンパク質(サブユニット)合成量の減少に加え,OXPHOSを構成するそれ以外のサブユニットの合成量も減少することがわかった.これらの核ゲノム由来のサブユニットは細胞質リボソームによって翻訳されるため,クロラムフェニコールによる影響を受けないはずである.しかし,これらはMTタンパク質と直接結合する相手であり,結合相手がいなくなったためフリーのプールはユビキチン化されプロテアソームによって分解される.一方で,MTタンパク質と直接結合しないOXPHOSサブユニットの合成量はほとんど変化しない.やや横道にそれるが,この研究を含め過去の研究18, 25)からも,ある遺伝子をノックアウトするとその翻訳産物のみならず結合相手はオーファン(孤児)となり,分解されうることが示唆されている.したがって遺伝子のノックアウトは単にその遺伝子から発現するタンパク質の存在がなくなるだけでなく,結合パートナーにも影響を及ぼすことも考慮すべきであろう.
3)短時間パルス標識型のpSILAC法
pSILAC法では,少なくとも4時間程度パルス標識しないと,安定同位体アミノ酸で標識された新生タンパク質は未標識タンパク質に埋もれてしまい質量分析で検出できない.したがって,pSILAC法はできたてほやほやのタンパク質を捉えることはできない.合成された直後の新生タンパク質を定量するために,tandem mass tag(TMT)標識26)とpSILAC法を組み合わせることで,15分~2時間のパルス標識時間で合成されるタンパク質を定量できる手法が開発されている27, 28)(図2B).TMT試薬はいわゆるサンプルのバーコーディング(多重化)ができ,複数サンプルを含む混合サンプルを同時に測定・定量することができる.TMT試薬はレポーター・スペーサー・アミン反応基の3部から構成され,レポーターもスペーサーも各々の分子構造は同じであるが一部の原子が安定同位体に置換されているため,TMT試薬全体の質量が変わらないように調整されている.したがって,異なる試料由来の消化ペプチドをレポーターの質量が異なるTMT試薬でそれぞれ標識・混合後に測定しても,ペプチドの質量は変わらずMS1レベルでは単一のピークとして検出される.一方,MS/MSでレポーター部が開裂することで,各試料由来の異なる質量のレポーターイオンが観測され,試料間の相対定量が可能となる.
話を元に戻すと,15分間~2時間のみHeavyアミノ酸でパルス標識した細胞では,Heavy標識された新生タンパク質の量はごくわずかであるが,Heavyアミノ酸で完全標識した細胞から得た消化ペプチドをTMT標識し,それを“シグナルブースティング”用にスパイクインすることで,付随的に極微量の新生タンパク質も定量できる(図2B).この手法の応用例としてたとえば,ストレスによってmTORC1またはeIF2αのリン酸化を介した翻訳抑制が誘導されることが知られているが,本法を用いることで両経路は共通のタンパク質の翻訳を抑制することが明らかにされている27).
4)AHAを用いたパルス標識
アジドホモアラニン(L-azidohomoalanine:AHA)29)はメチオニンの構造類似体であり(図2C),アジド基を有するためクリック反応によりアルキンビーズで精製することができる.安定同位体アミノ酸と同様にAHA存在下で細胞を培養することで(この場合メチオニンフリーの培地を使用),新生タンパク質を標識でき,かつクリック反応により精製できる特長がある.したがって,短時間(1~2時間)のパルス標識で得られた微量の新生タンパク質に対しても濃縮することで,質量分析による定量が可能である30–32).また,蛍光分子アルキンを用いることでフローサイトメトリー33)等を検出器として利用できる.有糸分裂期のタンパク質合成を単一細胞レベルで定量することで,有糸分裂期でも活発にタンパク質合成が行われていることもわかってきた34–36).
5)ピューロマイシンを用いたパルス標識
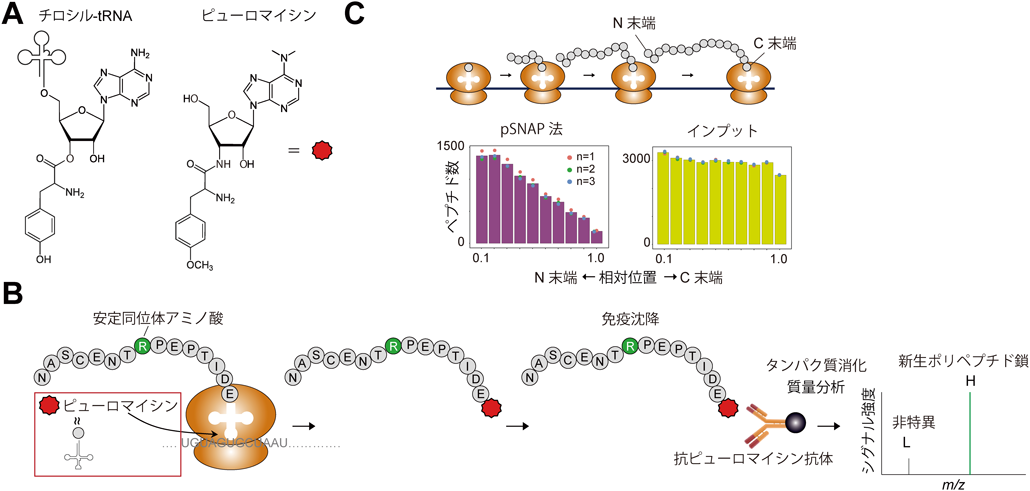
上述の安定同位体アミノ酸やAHAを用いる手法は,新しく合成された(翻訳後の)新生タンパク質プールを検出できるものの,リボソームで伸長中の新生ポリペプチド鎖を検出することは困難である.ピューロマイシンはアミノアシルtRNAの構造類似体であるため,リボソームで合成中の新生ポリペプチド鎖のC末端に取り込まれる(図3A).ピューロマイシンが結合した新生ポリペプチド鎖はそれ以上伸長することができず,リボソームから脱離する.このピューロマイシン標識ポリペプチドを生化学的に精製し質量分析することで,新生鎖の大規模計測が可能である.
PUNCH-P法(puromycin-associated nascent chain proteomics)37)では超遠心で精製したリボソームとビオチンピューロマイシンを試験管内で反応させ,ビオチン標識した新生ポリペプチド鎖をストレプトアビジンビーズで精製し,プロテオーム解析に供する.また,O-プロパルギル-ピューロマイシン(O-propargyl-puromyin:OPP)は分子内にアルキンを有するため,上述のAHAと同様にOPP標識ポリペプチド鎖にクリック反応を介してビオチンアジドでビオチンを付加し,ストレプトアビジンビーズで精製することができる38, 39).さらに,OPPは細胞膜透過性を有するため細胞内で新生ポリペプチド鎖を標識できる.一方,ビオチン–ストレプトアビジンなどの強固な結合に基づきアフィニテイ精製する際は,SDS等を用いて“激しい”条件でビーズを洗浄できるが,それでも非特異的なタンパク質の混入は避けられないことがこれまでのタンパク質相互作用プロテオミクスから明らかにされている31, 40, 41).そこで,我々は真の新生ポリペプチド鎖と非特異的なタンパク質を区別可能なpSNAP法(puromycin- and SILAC labeling-based nascent polypeptidome profiling)を開発した42)(図3B).この方法では,ピューロマイシンに加え安定同位体アミノ酸(アルギニンとリシン)も培地に加え,30分~2時間程度パルス標識をする.新生ポリペプチド鎖のC末端はピューロマイシンで標識され,さらに安定同位体アミノ酸によっても二重に標識される.その後,抗ピューロマイシン抗体を用いて,二重標識ポリペプチド鎖を精製し,質量分析する.真の新生ポリペプチド鎖であれば安定同位体アミノ酸で標識されたペプチドとして検出され,非特異的なタンパク質であれば未標識ペプチドとして検出されるので両者を区別できる(図3B).リボソームは新生ポリペプチド鎖をN末端側からC末端側にかけて伸長するため,新生ポリペプチド鎖を濃縮できていればN末端側由来のペプチドが多く同定されるはずである.実際に,安定同位体アミノ酸で標識されたペプチドはタンパク質配列内のN末端側に偏っており(図3C),この結果は本法で新生ポリペプチド鎖の大規模解析が可能であることを意味している.
6)ポリペプチジルtRNAの包括的解析
上述の安定同位体アミノ酸やAHA,ピューロマイシンといった標識剤を用いず,翻訳中間体のポリペプチジルtRNAを精製しプロテオーム解析する手法も最近開発されている43).RNAと架橋したタンパク質の濃縮法44)と同様に,細胞内のポリペプチジルtRNAをまず液液抽出により濃縮し,その後シリカカラムで単離する.ポリペプチジルtRNAのポリペプチド部分は,高pH・高温条件下で切断し,トリプシン消化物を質量分析する.本法は,標識が不要で翻訳時のスナップショット産物をプロファイルできる特長を持つ.さらに原理的には培養細胞から組織サンプルに対しても幅広く適用できる特長を持つ優れた方法である.
4. 新生タンパク質・ポリペプチド鎖の修飾とタンパク質の量的制御
1)新生タンパク質の修飾プロファイル
上述のパルス標識法と修飾ペプチド濃縮法を組み合わせることで,新生タンパク質特異的に起こる修飾を捉えることも可能になってきた45–47).原理としてはpSILAC法により新生タンパク質を標識し,その後,修飾ペプチド濃縮技術によりリン酸化ペプチド等を濃縮した画分と,非修飾ペプチドの画分をそれぞれ質量分析する.この方法により,リン酸化はタンパク質の一生のうち後半に起きる傾向が捉えられている46).一方で,新生タンパク質に選択的に起きるリン酸化も同定されている.たとえば,プロテアソームサブユニットPSMA5の新生タンパク質へのリン酸化は,PSMA5の正しいフォールディングに必要なタンパク質複合体アセンブリ中間体である可能性が示唆されている46).リン酸化のみならずアセチル化やユビキチン化もそれらの修飾に対する抗体を用いることで,プロテオームワイドに若いタンパク質と古いタンパク質間の修飾状態を測定できる47).たとえば,新生タンパク質由来のユビキチン化ペプチドは分解が促進される傾向にあり,これはユビキチン化タンパク質の約30%は合成された直後のタンパク質上で優先的にユビキチン化が起きること48–50)と一致する.
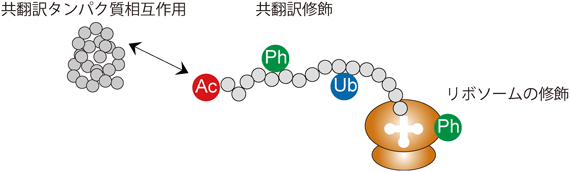
2)新生ポリペプチド鎖の修飾プロファイル
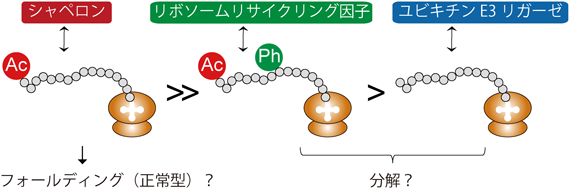
上述のいずれの方法もリボソームで翻訳中の新生ポリペプチド鎖に起きる修飾(共翻訳修飾)を捉えることは困難である.筆者らが開発したpSNAP法42)を用いることで,プロテオームワイドとはいえないが新生ポリペプチド鎖に起きるリン酸化やアセチル化を捉えることに成功している.たとえば,G3BP1の149番目のセリンやHSP90AA1の231番目のセリン等のリン酸化は新生ポリペプチド鎖レベルで修飾されており,過去の報告51, 52)とも一致する.興味深いことに,ヒストンH1.5のN末端由来の新生ポリペプチド鎖としてN末端アセチル化型,N末端アセチル化とリン酸化型,未修飾型の3種が検出され,存在量もこの順番で減少していた(図4).これらの修飾の意義を理解するために,未修飾もしくは修飾を含む合成ペプチドとHeLa細胞ライセートを用いたプルダウンアッセイを行い,結合タンパク質を質量分析により同定した.その結果,未修飾型には複数のユビキチンE3リガーゼが結合した.これは,未修飾型は不安定であるためユビキチン–プロテアソームによって分解されることを示唆しており,3種のペプチドフォームのうち未修飾型が細胞内での存在量が最も低いことと一致する.一方で,N末端アセチル化型はシャペロンと結合し,またアセチル化によって安定化53–55)されたため最も存在量が高かったと考えられる.N末端アセチル化とリン酸化型はリボソームリサイクリング因子PELOTA56)と選択的に結合した.これは異常な新生鎖を検知しリン酸化修飾することで(たとえば,異常な構造変化によりその部位がリン酸化修飾されうる),PELOTAやその関連因子により異物を除去・監視するシステムを有しているのかもしれない.この仮説はさらなる検証が必要であるが,この修飾型は3種のフォームの中で中間の存在量を示すこととも一致する.今後,アセチル化により安定化される機構や逆に未修飾であれば不安定化される機構に関して,構造生物学や生化学を駆使したメカニズムの解明が必須である.
以上のような若いタンパク質と古いタンパク質間での翻訳後修飾の質と量の計測は始まったばかりであるが,タンパク質恒常性維持のための新たな制御機構が隠されている可能性もあると筆者は考えている.
引用文献References
1) Schwanhäusser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., Chen, W., & Selbach, M. (2011) Global quantification of mammalian gene expression control. Nature, 473, 337–342.
2) Lubeck, E. & Cai, L. (2012) Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods, 9, 743–748.
3) Battich, N., Stoeger, T., & Pelkmans, L. (2013) Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods, 10, 1127–1133.
4) Kristensen, A.R., Gsponer, J., & Foster, L.J. (2013) Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol., 9, 689.
5) Morisaki, T., Lyon, K., DeLuca, K.F., DeLuca, J.G., English, B.P., Zhang, Z., Lavis, L.D., Grimm, J.B., Viswanathan, S., Looger, L.L., et al. (2016) Real-time quantification of single RNA translation dynamics in living cells. Science, 352, 1425–1429.
6) Wu, B., Eliscovich, C., Yoon, Y.J., & Singer, R.H. (2016) Translation dynamics of single mRNAs in live cells and neurons. Science, 352, 1430–1435.
7) Yan, X., Hoek, T.A., Vale, R.D., & Tanenbaum, M.E. (2016) Dynamics of translation of single mRNA molecules in vivo. Cell, 165, 976–989.
8) Wang, C., Han, B., Zhou, R., & Zhuang, X. (2016) Real-time imaging of translation on single mRNA transcripts in live cells. Cell, 165, 990–1001.
9) Ingolia, N.T., Lareau, L.F., & Weissman, J.S. (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell, 147, 789–802.
10) Schwanhäusser, B., Wolf, J., Selbach, M., & Busse, D. (2013) Synthesis and degradation jointly determine the responsiveness of the cellular proteome. BioEssays, 35, 597–601.
11) Beyer, A., Hollunder, J., Nasheuer, H.-P., & Wilhelm, T. (2004) Post-transcriptional expression regulation in the yeast Saccharomyces cerevisiae on a genomic scale. Mol. Cell. Proteomics, 3, 1083–1092.
12) Semenza, G.L. & Wang, G.L. (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol., 12, 5447–5454.
13) Buccitelli, C. & Selbach, M. (2020) mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet., 21, 630–644.
14) Liu, Y., Beyer, A., & Aebersold, R. (2016) On the dependency of cellular protein levels on mRNA abundance. Cell, 165, 535–550.
15) Fortelny, N., Overall, C.M., Pavlidis, P., & Freue, G.V.C. (2017) Can we predict protein from mRNA levels? Nature, 547, E19–E20.
16) Franks, A., Airoldi, E., & Slavov, N. (2017) Post-transcriptional regulation across human tissues. PLOS Comput. Biol., 13, e1005535.
17) Upadhya, S.R. & Ryan, C.J. (2022) Experimental reproducibility limits the correlation between mRNA and protein abundances in tumor proteomic profiles. Cell Rep. Methods, 2, 100288.
18) Taggart, J.C., Zauber, H., Selbach, M., Li, G.-W., & McShane, E. (2020) Keeping the proportions of protein complex components in check. Cell Syst., 10, 125–132.
19) Nusinow, D.P., Szpyt, J., Ghandi, M., Rose, C.M., McDonald, E.R. 3rd, Kalocsay, M., Jané-Valbuena, J., Gelfand, E., Schweppe, D.K., Jedrychowski, M., et al. (2020) Quantitative proteomics of the cancer cell line encyclopedia. Cell, 180, 387–402.e16.
20) Jovanovic, M., Rooney, M.S., Mertins, P., Przybylski, D., Chevrier, N., Satija, R., Rodriguez, E.H., Fields, A.P., Schwartz, S., Raychowdhury, R., et al. (2015) Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science, 347, 1259038.
21) Iwasaki, S. & Ingolia, N.T. (2017) The growing toolbox for protein synthesis studies. Trends Biochem. Sci., 42, 612–624.
22) Selbach, M., Schwanhäusser, B., Thierfelder, N., Fang, Z., Khanin, R., & Rajewsky, N. (2008) Widespread changes in protein synthesis induced by microRNAs. Nature, 455, 58–63.
23) Baek, D., Villén, J., Shin, C., Camargo, F.D., Gygi, S.P., & Bartel, D.P. (2008) The impact of microRNAs on protein output. Nature, 455, 64–71.
24) Imami, K., Selbach, M., & Ishihama, Y. (2023) Monitoring mitochondrial translation by pulse SILAC. J. Biol. Chem., 299, 102865.
25) Padovani, C., Jevtić, P., & Rapé, M. (2022) Quality control of protein complex composition. Mol. Cell, 82, 1439–1450.
26) Thompson, A., Schäfer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., Neumann, T., Johnstone, R., Mohammed, A.K., & Hamon, C. (2003) Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem., 75, 1895–1904.
27) Klann, K., Tascher, G., & Münch, C. (2020) Functional translatome proteomics reveal converging and dose-dependent regulation by mTORC1 and eIF2α. Mol. Cell, 77, 913–925.e4.
28) Schäfer, J.A., Bozkurt, S., Michaelis, J.B., Klann, K., & Münch, C. (2022) Global mitochondrial protein import proteomics reveal distinct regulation by translation and translocation machinery. Mol. Cell, 82, 435–446.e7.
29) Kiick, K.L., Saxon, E., Tirrell, D.A., & Bertozzi, C.R. (2002) Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. USA, 99, 19–24.
30) Dieterich, D.C., Link, A.J., Graumann, J., Tirrell, D.A., & Schuman, E.M. (2006) Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA, 103, 9482–9487.
31) Eichelbaum, K., Winter, M., Berriel Diaz, M., Herzig, S., & Krijgsveld, J. (2012) Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol., 30, 984–990.
32) Schanzenbächer, C.T., Sambandan, S., Langer, J.D., & Schuman, E.M. (2016) Nascent proteome remodeling following homeostatic scaling at hippocampal synapses. Neuron, 92, 358–371.
33) Sun, R., Cheng, E., Velásquez, C., Chang, Y., & Moore, P.S. (2019) Mitosis-related phosphorylation of the eukaryotic translation suppressor 4E-BP1 and its interaction with eukaryotic translation initiation factor 4E (eIF4E). J. Biol. Chem., 294, 11840–11852.
34) Imami, K., Milek, M., Bogdanow, B., Yasuda, T., Kastelic, N., Zauber, H., Ishihama, Y., Landthaler, M., & Selbach, M. (2018) Phosphorylation of the ribosomal protein RPL12/uL11 affects translation during mitosis. Mol. Cell, 72, 84–98.e9.
35) Imami, K. & Yasuda, T. (2019) Measuring protein synthesis during cell cycle by Azidohomoalanine (AHA) labeling and flow cytometric analysis. Bio Protoc., 9, e3215.
36) Shuda, M., Velásquez, C., Cheng, E., Cordek, D.G., Kwun, H.J., Chang, Y., & Moore, P.S. (2015) CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc. Natl. Acad. Sci. USA, 112, 5875–5882.
37) Aviner, R., Geiger, T., & Elroy-Stein, O. (2013) Novel proteomic approach (PUNCH-P) reveals cell cycle-specific fluctuations in mRNA translation. Genes Dev., 27, 1834–1844.
38) Forester, C.M., Zhao, Q., Phillips, N.J., Urisman, A., Chalkley, R.J., Oses-Prieto, J.A., Zhang, L., Ruggero, D., & Burlingame, A.L. (2018) Revealing nascent proteomics in signaling pathways and cell differentiation. Proc. Natl. Acad. Sci. USA, 115, 2353–2358.
39) Uchiyama, J., Ishihama, Y., & Imami, K. (2021) Quantitative nascent proteome profiling by dual-pulse labelling with O-propargyl-puromycin and stable isotope-labelled amino acids. J. Biochem., 169, 227–236.
40) Howden, A.J.M., Geoghegan, V., Katsch, K., Efstathiou, G., Bhushan, B., Boutureira, O., Thomas, B., Trudgian, D.C., Kessler, B.M., Dieterich, D.C., et al. (2013) QuaNCAT: Quantitating proteome dynamics in primary cells. Nat. Methods, 10, 343–346.
41) Mellacheruvu, D., Wright, Z., Couzens, A.L., Lambert, J.P., St-Denis, N.A., Li, T., Miteva, Y.V., Hauri, S., Sardiu, M.E., Low, T.Y., et al. (2013) The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods, 10, 730–736.
42) Uchiyama, J., Roy, R., Wang, D.O., Morikawa, K., Kawahara, Y., Iwasaki, M., Yoshino, C., Mishima, Y., Ishihama, Y., & Imami, K. (2022) pSNAP: Proteome-wide analysis of elongating nascent polypeptide chains. iScience, 25, 104516.
43) Yamakawa, A., Niwa, T., Chadani, Y., Kobo, A., & Taguchi, H. (2023) A method to enrich polypeptidyl-tRNAs to capture snapshots of translation in the cell. Nucleic Acids Res., 51, gkac1276.
44) Trendel, J., Schwarzl, T., Horos, R., Prakash, A., Bateman, A., Hentze, M.W., & Krijgsveld, J. (2019) The human RNA-binding proteome and its dynamics during translational arrest. Cell, 176, 391–403.e19.
45) Wu, C., Ba, Q., Lu, D., Li, W., Salovska, B., Hou, P., Mueller, T., Rosenberger, G., Gao, E., Di, Y., et al. (2021) Global and site-specific effect of phosphorylation on protein turnover. Dev. Cell, 56, 111–124.e6.
46) Hammarén, H.M., Geissen, E.-M., Potel, C., Beck, M., & Savitski, M.M. (2022) Protein-peptide turnover profiling reveals wiring of phosphorylation during protein maturation. Nat. Commun., 13, 7431.
47) Zecha, J., Gabriel, W., Spallek, R., Chang, Y.C., Mergner, J., Wilhelm, M., Bassermann, F., & Kuster, B. (2022) Linking post-translational modifications and protein turnover by site-resolved protein turnover profiling. Nat. Commun., 13, 165.
48) Kim, W., Bennett, E.J., Huttlin, E.L., Guo, A., Li, J., Possemato, A., Sowa, M.E., Rad, R., Rush, J., Comb, M.J., et al. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell, 44, 325–340.
49) Schubert, U., Antón, L.C., Gibbs, J., Norbury, C.C., Yewdell, J.W., & Bennink, J.R. (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature, 404, 770–774.
50) Vabulas, R.M. & Hartl, F.U. (2005) Protein synthesis upon acute nutrient restriction relies on proteasome function. Science, 310, 1960–1963.
51) Reineke, L.C., Tsai, W.C., Jain, A., Kaelber, J.T., Jung, S.Y., & Lloyd, R.E. (2017) Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol., 37, e00596-16.
52) Backe, S.J., Sager, R.A., Woodford, M.R., Makedon, A.M., & Mollapour, M. (2020) Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem., 295, 11099–11117.
53) Varland, S., Silva, R.D., Kjosås, I., Faustino, A., Bogaert, A., Billmann, M., Boukhatmi, H.. Kellen, B., Costanzo, M., Drazic, A., Osberg, C., et al. (2022) N-terminal acetylation shields proteins from degradation and promotes age-dependent motility and longevity. bioRxiv. 2022.09.01.505523 doi:10.1101/2022.09.01.505523
54) Linster, E., Forero Ruiz, F.L., Miklankova, P., Ruppert, T., Mueller, J., Armbruster, L., Gong, X., Serino, G., Mann, M., Hell, R., et al. (2022) Cotranslational N-degron masking by acetylation promotes proteome stability in plants. Nat. Commun., 13, 810.
55) Mueller, F., Friese, A., Pathe, C., da Silva, R.C., Rodriguez, K.B., Musacchio, A., & Bange, T. (2021) Overlap of NatA and IAP substrates implicates N-terminal acetylation in protein stabilization. Sci. Adv., 7, eabc8590.
56) Pisareva, V.P., Skabkin, M.A., Hellen, C.U.T., Pestova, T.V., & Pisarev, A.V. (2011) Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J., 30, 1804–1817.
57) Ingolia, N.T., Ghaemmaghami, S., Newman, J.R.S., & Weissman, J.S. (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science, 324, 218–223.