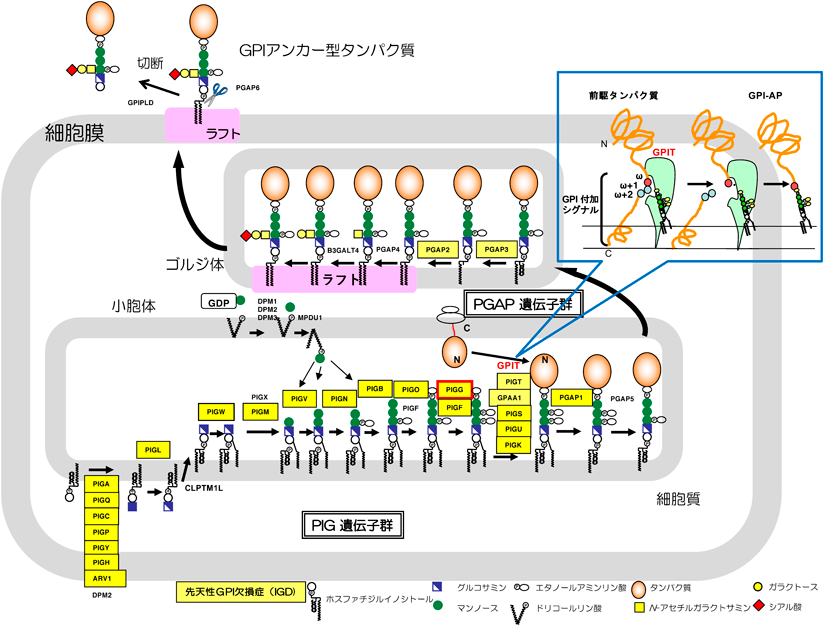
GPIアンカー型タンパク質(glycosylphosphatidylinositol anchored protein:GPI-AP)は前駆タンパク質とGPIアンカー部分が小胞体で別々に合成され,PIGKを触媒因子とする五つの因子(PIGK, PIGS, PIGT, PIGU, GPAA1)からなるGPIトランスアミダーゼ(GPIT)複合体が前駆タンパク質のC末端のGPI付加シグナルを認識して切断し,GPIアンカーを付加する(図1).その後もGPIの糖鎖と脂質部分は小胞体とゴルジ体においてさまざまな修飾を受けて細胞表面のラフトと呼ばれるドメインに運ばれる.ヒトや哺乳動物では150種以上のGPI-APが知られており,それぞれ酵素,受容体,接着因子,補体制御因子などとして機能し,受精や形態形成,神経発達,免疫などにおいて重要な役割を担っている1).
GPIアンカーの構造は1980年代後期にラットThy-1とトリパノソーマのvariant surface glycoproteinで決定された.GPIのコア構造はEthN-P-6-Man-α1,2-Man-α1,6-Man-α1,4-GlcN-α1,6-myoinositol-phospholipid(GlcN:グルコサミン,Man:マンノース,EthN-P:エタノールアミンリン酸)で,この三つ目のMan(Man3)に結合するEthN-Pのアミノ基を介して前駆タンパク質のカルボキシ末端に付加されることから,このEthN-Pは“bridging EthN-P”と呼ばれている.このGPIのコア構造はすべての真核生物で保存されている1).
GPI-APの生合成経路ではGPIがタンパク質に付加されるまでに関与する遺伝子はPIGA, PIGBなどPIG(phosphatidylinositol glycan)遺伝子群としてPIG+アルファベット,の名前がついている.その後の修飾に関与する遺伝子をPGAP(post GPI attachment to proteins)遺伝子群として1から6までの数字で示し,合計30個の遺伝子が関わっている(図1)1–3).これらの遺伝子の変異により,GPI-APの発現低下や種々の構造異常が起こり,精神運動発達の遅れやてんかんを主症状とする先天性グリコシルホスファチジルイノシトール(GPI)欠損症(inherited GPI deficiency:IGD)を発症する.重症例では顔貌異常,手指・足趾末端骨の形成不全,聴力・視力の異常,ヒルシュスプルング病や横隔膜ヘルニアなどの内臓奇形を伴う4).潜性遺伝性の疾患である.PIGA欠損症(PIGA-IGD)については,PIGA遺伝子がX染色体上にあるので,無症状のヘテロの母親から生まれた男児のみが罹患する5).GPIの完全欠損は胎生致死になることから,IGD患者の多くは活性が低下する部分欠損症である.末梢血のフローサイトメトリーにより顆粒球上に発現するGPI-APであるCD16bの発現レベルが低下することでスクリーニングが可能6)で,現在国内外で約500例の報告がある.
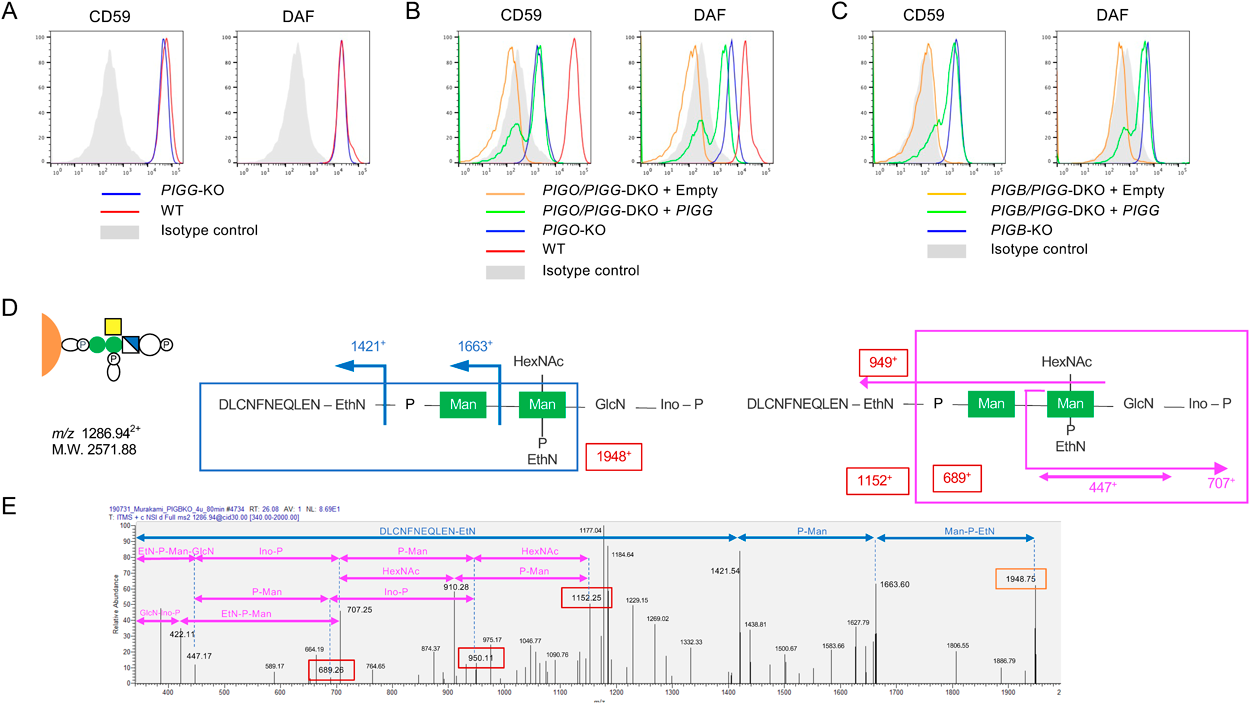
生合成遺伝子のうち,PIGGはGPIのMan2にEthN-Pを付加する酵素であるが,このEth-Pは小胞体においてGPIが前駆タンパク質に付加されたのちに,PGAP5によって速やかに除かれる.すなわちPIGGノックアウト(knockout:KO)細胞ではCD59やdecay-accelerating factor(DAF)などのGPI-APは正常細胞と同様に脂質部分の修飾を受けて正常構造,正常レベルで細胞表面に発現するので,このPIGGのステップの生理的な役割が不明であった(図1,図2A)1).
ところがPIGG欠損症の患者が次々と報告され,難治性てんかんや精神発達の遅れなど重篤な神経症状を示すことから,PIGGが担うステップが神経発達に重要であることが明らかになった7).
前述したようにGPIのコア構造はすべての真核生物で保存されていると長年考えられてきたが,我々の研究によりそのドグマを覆す新規の構造が見つかり,それがPIGG欠損症(PIGG-IGD)の発症メカニズムを解く鍵になった.
GPI生合成遺伝子のうち,Man3に“bridging EthN-P”を付加するPIGOやMan3を付加するPIGBのKO細胞ではGPI-APであるCD59の発現が10%程度残る.これらにさらにMan2にEthN-Pを付加するPIGGをKOしてダブルKO(DKO)細胞を作製するとCD59は完全に欠損する(図2B, C).この結果によりKO細胞で残存しているCD59の発現はPIGGが付加するMan2のEthN-Pを介したものであることが予想された.
そこでHEK293細胞に四重タグを付加したHis-FLAG-GST-FLAG(HFGF)-CD59を高発現させ,さらにPIGBをKOした.野生型細胞とPIGBKO細胞で発現するHFGF-CD59の構造を比較するため,GPI部分をPI特異的ホスホリパーゼC(PIPLC)で切断して可溶化後グルタチオンビーズで精製し,SDS-PAGEののちトリプシンによるin-gel消化を行い,LC(C18カラム)-ESI-MSでGPIの糖鎖構造を解析した.PIGBKO細胞由来のサンプルでは,CD59のC末端の11アミノ酸に2個のManとHexNAcの側鎖がつながったものやさらにHexがつながったprecursorイオンを検出し,そのMS/MS解析によりGPIの診断イオン(m/z 422+,447+,707+,1421+,1663+)とともに,Man2のEthN-Pを介してCD59のペプチドが結合しているフラグメント,P-Man-(EthN-P-)Man-GlcN(m/z 689+)とpeptide-EthN-P-Man-(EthN-P-)Man(m/z 1948+)を検出した(図2D, E).すなわちPIGBKO細胞では予想どおり,Man2のEthN-Pを介してCD59が結合していることが明らかになった.さらに野生型の細胞に発現するHFGF-CD59のGPIの構造を詳細に解析したところ,約10%のprecursorイオンでこの新規構造が検出された8).
上記の結果から,神経組織には主としてこの新規の構造で発現するタンパク質が存在し,PIGG欠損症ではそのようなタンパク質の発現が減少することにより神経発達の異常が生じていると考え,野生型細胞と比べてPIGG欠損細胞で特異的に発現が低下しているタンパク質を指標に,新規構造で発現するタンパク質を検索した.
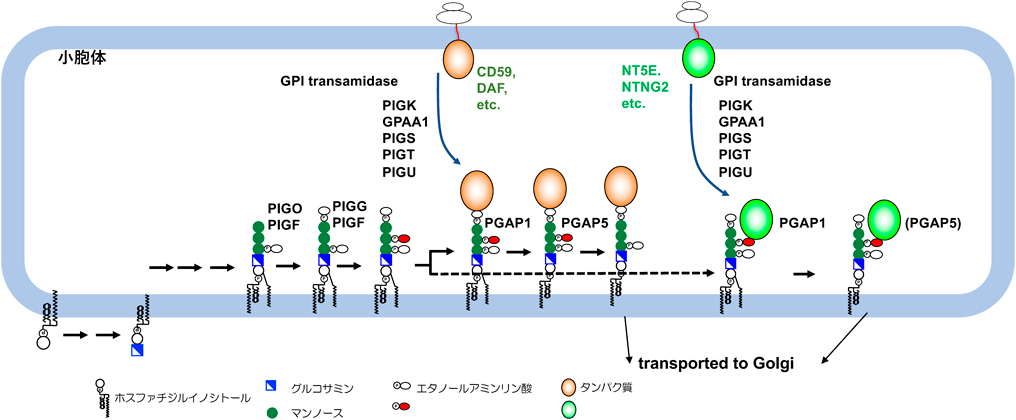
HEK293細胞のPIGGKO細胞と野生型の細胞の抽出液からTriton X-114による相分離で膜タンパク質を抽出し,さらにPIPLC処理により水相に移ったタンパク質の半定量プロテオミクスを行い,検出された36個のGPI-APのうちPIGGKOで有意に減少している7個のGPI-APを選択した.また,UniProt(https://www.uniprot.org/)にて神経組織で発現している50個のGPI-APを選び,そのcDNAをPIGGKO細胞に遺伝子導入してPIGGをレスキューした細胞とフローサイトメトリーにより発現量を比べると18個のGPI-APがPIGGKOで減少していた.これらの中からCD73(NT5E)とNetrinG2(NTNG2)を選び,質量分析によるGPIの解析を行った.N末端にHAタグを付加したそれぞれのcDNAをExpi 293F細胞に遺伝子導入し,anti-HAビーズで精製後SDS-PAGEを施行し,NT5EはトリプシンでNTNG2はキモトリプシンでin gel消化を行い,前者はhydrophilic(HILIC)カラム,後者はC18カラムによるLC-ESI-MSでGPIの構造を解析した.NT5EではC末端の2アミノ酸(FS)に,NTNG2ではC末端の22個のアミノ酸(TGVRCEQPRCDPADDDGGLDCD)に,3個のManとHexNAcの側鎖がつながったものやさらにHexがつながったprecursorイオンを検出し,両者ともそのMS/MS解析によりGPIの診断イオンとともに,Man2のEthN-Pを介してペプチドが結合しているフラグメントを検出した8).また検出できたすべてのprecursorイオンについて,この新規構造が検出された.すなわちCD73(NT5E)とNetrinG2については,Man2のEthN-Pを介してタンパク質が結合している新規構造をとることがわかった.上記の結果から神経細胞に発現するタンパク質のなかにはCD73やNetrinG2のように主としてこの新規構造で発現するタンパク質があり,PIGG欠損症ではこれらのタンパク質の発現が低下し,神経症状が生じていると考えられた(図3).
4. GPIトランスアミダーゼ(GPIT)について
前駆タンパク質にGPIを付加するGPITはどのように前駆タンパク質を区別して,GPIのMan2のEth-Pを介して付加しているのだろうか.2021年に,AlphaFold2を使って酵母のGPIT複合体の予想構造が発表された9)が,翌年実際に精製したヒトのGPIT複合体のクライオ電子顕微鏡を使った解析で,予想どおりの構造であることが証明された10, 11).その論文によると複合体に結合したGPIの一部,Man(EthN-P)-GlcN-palmitoylated phosphatidylinositolの構造がPIGKの触媒部位(H164, C206)の下部に検出され,そのアシル基がPIGUの膜貫通部位に張りつき,糖鎖はPIGT, PIGUの残基と水素結合していた11).一方前駆タンパク質のC末端のGPI付加シグナル(C-terminal signal peptides:CSP)は,決まったアミノ酸の配列ではなくPIGKによってC末端側で切断されるオメガサイトと呼ばれる側鎖の小さいアミノ酸(グリシン,アラニン,セリン,システイン,アスパラギン酸,アスパラギンのうちどれか)と10残基程度の親水性アミノ酸配列,10~20残基程度の疎水性アミノ酸配列からなる.上記の構造解析によりPIGKの触媒部位はちょうど小胞体膜から22 Å上部にあり,これはちょうどCSPの10残基程度の親水性アミノ酸の長さにほぼ一致するとともに,GPIコアの糖鎖部分の長さにも一致する11).つまり前駆タンパク質はC末端側の疎水性アミノ酸で小胞体膜に埋め込まれ,10残基程度の親水性アミノ酸が水素結合によりPIGKの触媒部位周辺の残基と結合し,オメガサイトが触媒部位上部に結合して切断され,そばにいるGPIが結合する.新規構造を好んでとるCD73やNeteinG2の場合は,そのCSPとPIGK残基との結合の具合によってGPIとの位置関係が変化するのかもしれない.
5. 先天性PIGG欠損症(PIGG-IGD)について
PIGG-IGDの症状は,新規構造をとるCD73やNetrinG2など神経組織に高発現するGPI-APの発現低下によるものであると考えられる.実際にPIGG欠損症例では繊維芽細胞のCD73の発現低下が報告されている.CD73はアデノシン一リン酸(adenosine monophosphate:AMP)をアデノシンに変換する酵素であるが,最近の我々の研究で血清中の主なビタミンB2であるフラビンアデニンジヌクレオチド(flavin adenine dinucleotide:FAD)からフラビンモノヌクレオチド(flavin mononucleotide:FMN)への変換に必須の酵素であることがわかった12).FMNはGPI-APであるアルカリホスファターゼの働きで脱リン酸化されてリボフラビンになり初めて細胞内に取り込まれ,細胞内で再びリン酸化されて活性型となりミトコンドリアでの電子伝達系などのエネルギー産生に補酵素として働く.ビタミンB2としてFADのみを与えたGPI欠損細胞では細胞内ビタミンB2欠乏によりミトコンドリアの機能異常が起こる12).PIGG-IGDの重症例でもミトコンドリア機能異常が報告されている13).またNetrinG2欠損症の家系では,神経運動発達の遅れと筋緊張低下,自閉症様行動など,PIGG-IGDと共通する症状が報告されている14).
本研究の過程で,PIGO/PIGG DKO細胞にPIGGをレスキューすることで,フローサイトメトリーによるPIGG遺伝子変異の機能解析法を開発し(図2B),PIGG-IGDのphenotype-genotypeの解析が可能となった.PIGG-IGDでは多くのGPI-APの発現は低下しないので,他のIGDと異なり完全欠損の患者が報告されており,臓器奇形はなく難治性てんかんや小脳萎縮など神経系の重度の異常を主体とする13).一方,正常赤血球に高頻度に発現するEmm抗原という血液型抗原が知られており,その抗体所有者に誤って輸血し溶血を起こした例が国内でも報告されている.最近その抗体所有者がPIGGの完全欠損の患者であることが判明し,Emm抗原がMan2にEthN-PがついたフリーGPI(タンパク質が付加されていないGPI)であることが明らかになった.つまりPIGGの完全欠損症ではこの構造がないので,自然抗体を生じていた.これにより正常赤血球上にもフリーGPIが発現していることが明らかになった15).