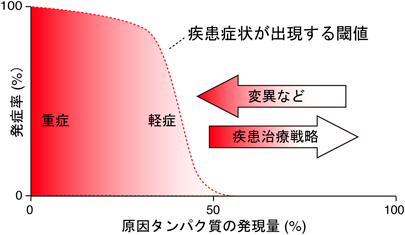
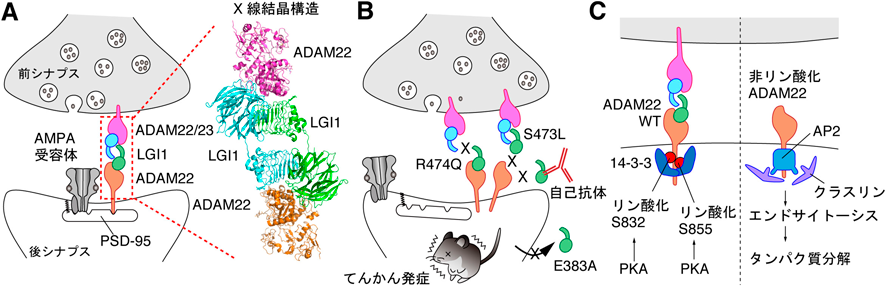
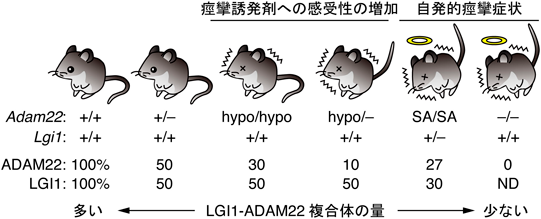
てんかん発症を抑制するためのLGI1–ADAM22タンパク質複合体の量的制御機構Regulatory mechanisms of the LGI1–ADAM22 protein levels to prevent epilepsy
1 自然科学研究機構生理学研究所分子細胞生理研究領域生体膜研究部門Division of Membrane Physiology, Department of Molecular and Cellular Physiology, National Institute for Physiological Sciences, National Institutes of Natural Sciences ◇ 〒444–8787 愛知県岡崎市明大寺町字東山5–1 ◇ 5–1 Higashiyama, Myodaiji-cho, Okazaki, Aichi 444–8787, Japan
2 総合研究大学院大学生命科学研究科生理科学専攻Department of Physiological Sciences, School of Life Science, SOKENDAI (The Graduate University for Advanced Studies) ◇ 〒444–8787 愛知県岡崎市明大寺町字東山5–1 ◇ 5–1 Higashiyama, Myodaiji-cho, Okazaki, Aichi 444–8787, Japan
3 名古屋大学大学院医学系研究科神経情報薬理学講座Department of Cell Pharmacology, Nagoya University Graduate School of Medicine ◇ 〒466–8550 愛知県名古屋市昭和区鶴舞町65 ◇ 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466–8550, Japan
発行日:2023年6月25日Published: June 25, 2023