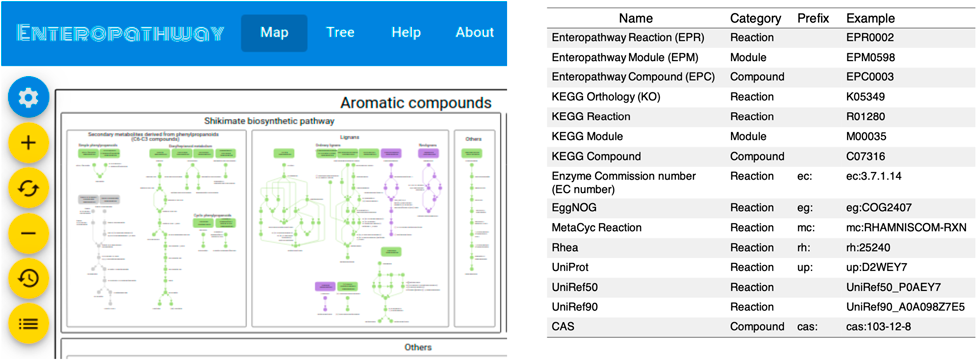
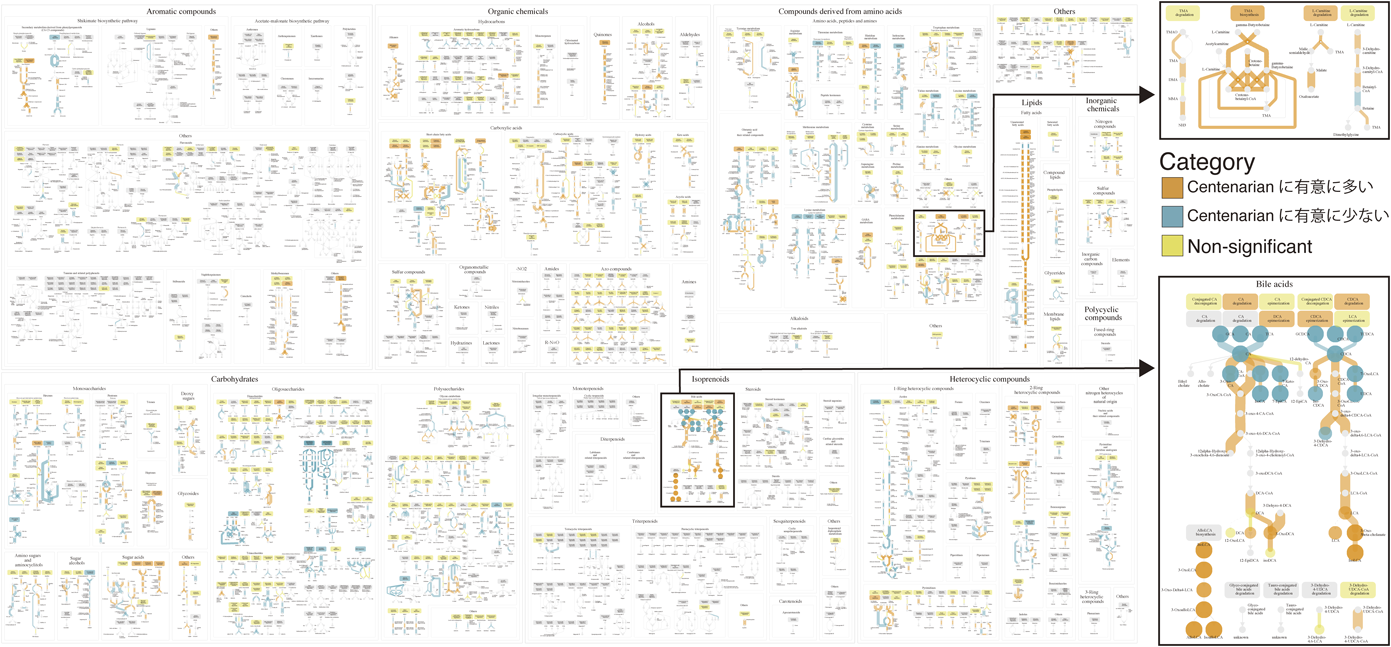
腸内細菌群集の網羅的代謝機能データベースFunctional annotation atlas for human gut microbiome
1 沖縄科学技術大学院大学統合オープンシステムユニットOkinawa Institute of Science and Technology ◇ 〒904–0495 沖縄県国頭郡恩納村字谷茶1919–1 ◇ 1919–1 Tancha, Onna-son, Kunigami-gun, Okinawa 904–0495, Japan
2 東京工業大学生命理工学院Tokyo Institute of Technology Department of Life Science and Technology ◇ 〒152–8550 東京都目黒区大岡山2–12–1 M6–4 ◇ 2–12–1 Ookayama, Meguro-ku, Tokyo 152–8550, Japan
発行日:2023年8月25日Published: August 25, 2023