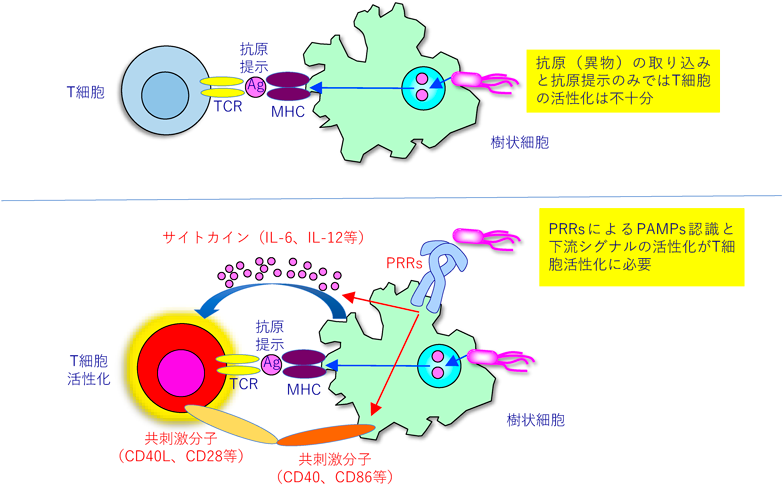
免疫系は大きく自然免疫と獲得免疫に分けられる.自然免疫は病原体感染初期に働く生体防御機構であり,マクロファージや樹状細胞を介した異物貪食,抗原提示,炎症誘導等を通して病原体排除に貢献する.一方,獲得免疫を担当するT細胞やB細胞は遺伝子再構成により生み出された抗原受容体を介して抗原認識を行い,その多様性や記憶が大きな特徴である1).この免疫系が発動する仕組みを解き明かしたのがJaneway Jr.であった.一般的に,タンパク質(抗原)の接種のみで,抗原特異的な抗体による体液性免疫や細胞傷害性T細胞による細胞性免疫といった獲得免疫は十分に誘導されず,アジュバントと呼ばれる死菌成分等との同時接種が必要であることが知られていた.実際,多くの研究者が,目的の抗体を得るためにマウスやウサギ等に免疫をする際に完全フロイントアジュバント(結核菌死菌を含む)を用いている.その作用機序は,抗原の安定化,B細胞の活性化,抗原提示の促進等によるものと漠然と考えられていた.筆者は免疫研究を開始した学部生のときに,指導教員に質問をしたが「経験的にアジュバントが必要なのだ」という返事が得られたのみであった.恥ずかしながら,筆者自身も深く考えていなかった.Janewayは,なぜアジュバントが必要なのか?そもそも免疫系は何をきっかけに発動するのか?という問いに対し,パターン認識受容体(pattern recognition receptors:PRRs)仮説を提唱した2).これは,我々の身体には病原体に固有に存在する病原体構成成分(pathogen-associated molecular patterns:PAMPs)を認識するPRRsが存在しており,この認識により開始される細胞応答が免疫系の発動であるというものである(図1).PAMPsとは,たとえば,細菌のリポ多糖(LPS)やペプチドグリカンといった,病原体に固有に存在しそれらの生育に必須の成分であるが,我々の身体には存在しない構成成分を指す.PRRsが実際に存在することを世界に先駆け発見したのが,Hoffmannであった3).1996年,Hoffmannは,ショウジョウバエにおいてTollと呼ばれる膜型タンパク質が真菌に対する抗菌ペプチドの産生と感染防御に必須であることをCell誌に報告した.遺伝子再構成系を持たず獲得免疫を有しないショウジョウバエでは,Tollを介して病原体の侵入を察知して生体防御を行うことが見いだされたのである.Tollは,ロイシンに富む細胞外領域と,インターロイキン1(IL-1)やIL-18受容体の細胞内領域に類似するドメイン[Toll-IL-1 receptor(TIR)ドメイン]から構成される膜型タンパク質である.一方,IL-1やIL-18受容体は細胞外にイムノグロブリン様領域を持ち,Tollの細胞外構造とは異なる.翌97年,Janewayは弟子であるMedzhitovとともにTollに似た分子がヒトにも存在していることをNatureに報告した4).彼らは,この分子をhTollと名づけ,過剰発現系の実験から,hTollがIL-1受容体やTollと同様にNF-κB経路を活性化することを見いだした.また,これにより標的遺伝子である炎症性サイトカインの発現を誘導することも示した.その後,hTollと類似構造を示す分子がヒトで10種類存在することがわかり,これらはToll-like receptor(TLR)ファミリーと呼ばれるようになった1).なお,hTollはTLR4である.
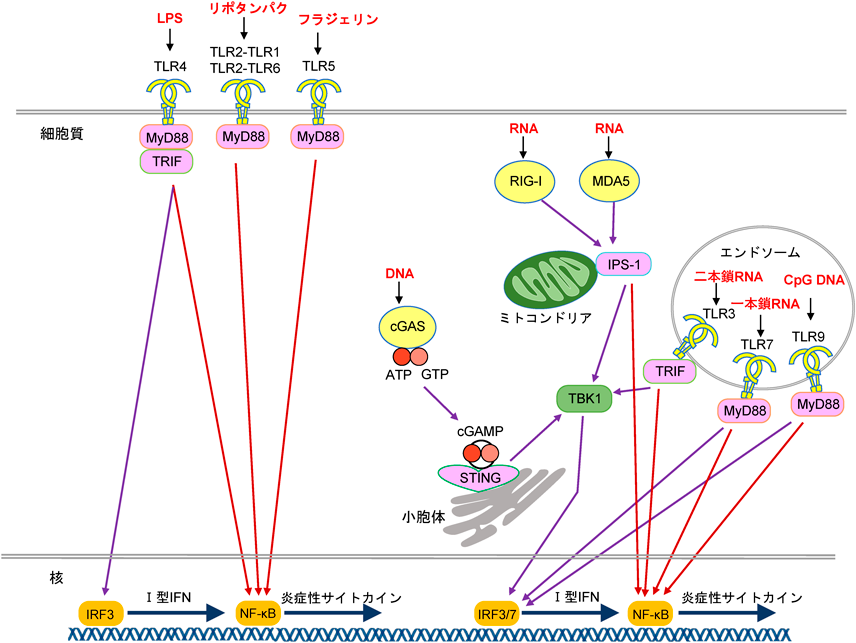
一方,筆者が大学院生として所属していた大阪大学・審良静男教授の研究室(当時は兵庫医科大学)では,MyD88と呼ばれる分子の解析を行っていた.MyD88はTIRドメインを有する細胞内因子であり,IL-1やIL-18受容体と会合しNF-κB経路活性化に必要な細胞内アダプター分子である5).興味深いことに,MyD88ノックアウトマウスは,LPS投与に対するショックに耐性を示しマクロファージからの炎症性サイトカインの産生も欠如していた6).このことから,MyD88はIL-1/IL-18受容体のみならずTLRファミリーの下流シグナル伝達分子であり,TLRのどれかがLPS受容体ではないかと考えられるようになった.そして,TLRファミリーの中でTLR4がLPS受容体であることがBeutlerや審良らによって発見された7, 8).その後,他のTLRファミリーのノックアウトマウスを用いた機能解析が審良研究室を中心に行われ,各TLRのリガンドが同定されるに至った(図2)1, 9, 10).TLR1, 2, 4, 5, 6, 10は細胞表面に局在しており,主に細菌表面の成分を認識する.TLR2-TLR1, TLR2-TLR6はリポタンパク質,TLR5はフラジェリンを認識する.一方,TLR3, 7, 8, 9, 13はエンドソームなどの細胞内オルガネラ膜に局在し,主に核酸を認識する1, 11, 12).TLR3は二本鎖RNA(double-stranded RNA:dsRNA),TLR7とTLR8は一本鎖RNA(single-stranded RNA:ssRNA),TLR9はDNAのメチル化されていないシトシンとグアニン(CpG)配列を認識する.TLRは主にマクロファージや樹状細胞といった抗原提示細胞に発現しており,PAMPs認識に伴いMyD88やMyD88類似分子であるTRIF等と会合し,下流へとシグナル伝達経路を活性化する.最終的に転写因子NF-κBやIRF(interferon regulatory factor)ファミリーを活性化することで,炎症性サイトカインやケモカインの分泌,共刺激因子の細胞表面への発現を増強させる1, 11, 12).こうした自然免疫応答は,獲得免疫の成立に大きく寄与する.したがって,TLRはアジュバント受容体ともいえる.TLRの発見により,長らく謎だったアジュバントの作用機序がわかったとともに,Janewayの仮説のとおり,PRRsによるPAMPs認識が免疫発動の起点となっていることが証明された.2011年のノーベル医学・生理学賞はTLRの発見に携わったHoffmannとBeutler,樹状細胞の発見者であるSteinmanに贈られた13).残念ながらPRRs概念の生みの親であり,hTollの発見も行ったJanewayは2003年に亡くなっていたため,対象とはならなかった.
TLRsは主にマクロファージや樹状細胞等の自然免疫担当細胞に発現している膜型受容体であり,細胞表面やエンドソームでPAMPsを認識する.一方,ウイルスの多くは上皮細胞等の非免疫細胞に感染し細胞質内で増殖する.このとき,細胞はウイルスRNAを細胞質内でTLR非依存的に感知し,抗ウイルス活性を持つI型インターフェロン(interferon:IFN)や炎症性サイトカインの発現を誘導することでウイルス複製を抑制する.このRNA認識に関わる因子として,藤田と米山によりRIG-I(retinoic acid-inducible gene-I)が発見された(図2)14).RIG-Iは,ATP加水分解酵素活性部位とRNAヘリカーゼドメイン,RNA結合ドメイン,下流へのシグナル伝達に必須であるCARD(caspase recruitment domain)を有している.RIG-Iと似た構造を示す分子としてMDA5が存在し,これらはRIG-I-like receptors(RLRs)と呼ばれる.RLRsはいずれもIFN誘導性遺伝子であり,免疫細胞以外の細胞にも広く発現している.RLRsがウイルスRNAを認識すると,ATP依存的に構造変化を起こし,CARDが露出することで,ミトコンドア膜上に局在するCARDを有するアダプター分子IPS-1(別名MAVS)と相互作用する15, 16).IPS-1はミトコンドリア膜上で凝集体を形成し,最終的にNF-κBやIRF3の活性化を介して炎症性サイトカインやI型IFNの産生を誘導する.RIG-Iは5′三リン酸短鎖二本鎖RNA, MDA5は長鎖二本鎖RNAを認識する.また,RIG-IはインフルエンザウイルスやC型肝炎ウイルス,MDA5は脳心筋炎ウイルスやSARS-CoV2の認識に関わる17, 18).一方,DNAウイルスなどに由来する細胞質内DNAの認識に関わる細胞内PRRsとしてcGAS(cyclic GMP-AMP synthase)が同定された(図2)19).cGASはさまざまな細胞において発現しており,細胞質内二本鎖DNAと結合することで,細胞内のGTPおよびATPからセカンドメッセンジャーとしての環状GMP-AMP(cyclic GMP-AMP:cGAMP)を合成する.cGAMPは,小胞体に局在するSTINGを介して下流にシグナルを伝達し,最終的にNF-κBやIRF3を活性化することで炎症性サイトカインやI型IFNの産生を誘導する20).
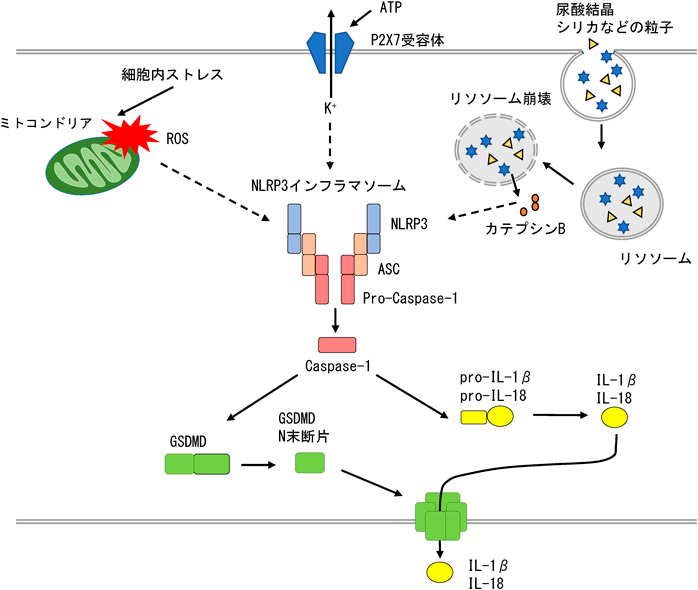
これら核酸認識に関わる細胞内PRRsに加え,細胞内PRRとしてNLRP3インフラマソームが知られている(図3)21).NLRP3は,尿酸結晶やシリカなどの粒子や細胞外ATP,細菌毒素などの因子により活性化される.NLRP3がこれらの刺激により活性化されると,アダプタータンパク質ASC(apoptosis-associated speck-like protein containing caspase recruitment domain)がリクルートされる.さらにASCとCaspase-1の前駆体が結合することで複合体が形成され,インフラマソームとなる.インフラマソームの形成によりCaspase-1の自己切断が誘導され,活性化型Caspase-1が生じる.活性化したCaspase-1はIL-1βやIL-18前駆体を成熟型へと切断するとともに,Gasdermin D(GSDMD)の切断も行う.切断されたGSDMDのN末端断片が細胞膜に孔を形成することで,細胞死(パイロトーシス)が誘導される.この細胞死に伴い,IL-1βやIL-18が細胞外へと放出され,免疫応答が惹起される.NLRP3は,PAMPsを直接認識し,インフラマソームを形成するのではなく,刺激によって誘導される活性酸素種(ROS)や酸化ミトコンドリアDNAの細胞質への流出,リソソームの崩壊,細胞内K+の流出などが引き金となる.NLRP3インフラマソームを活性化する細胞外ATPは,イオンチャネルであるP2X7受容体に作用することで細胞内K+の流出を生じさせる.また,尿酸結晶やシリカなどの粒子は細胞内に取り込まれるとリソソーム膜の破壊を引き起こし,カテプシンBなどのリソソーム内容物が細胞質に漏出することでNLRP3が活性化される.したがって,NLRP3インフラマソームは細胞障害を感知し炎症を惹起するセンサーとして機能していると考えられている21).また,NLRP3の活性型変異は,クライオピリン関連周期熱症候群(CAPS)で認められ,NLRP3インフラマソームの活性化が疾患と直接関連していることが示されている.これらの他,病原体の糖脂質認識に関わるC型レクチン受容体(C-type lectin receptors:CLRs)22)や,NLRP3に類似し細胞内でさまざまなPAMPs認識に関わるNod-like receptors(NLRs),DNA認識に関与するインフラマソーム構成因子であるAIM2-like receptors(ALRs)等が同定されている21).
PRRsはPAMPsだけでなく,損傷組織や壊死,パイロトーシスやネクロプトーシス等のシグナル伝達経路で制御された細胞死を起こした細胞から放出される内因性因子も認識し炎症応答を誘導する.このような内因性因子は,ダメージ関連分子パターン(damage-associated molecular patterns:DAMPs)あるいは危険信号(danger signal)と呼ばれる11, 23).非ヒストン核タンパク質であるHMGB1(high mobility group protein B1)やミトコンドリア由来分子,自己核酸,ATP,尿酸結晶などの分子がDAMPsとして作用する.HMGB1はTLR2, TLR4,ミトコンドリアDNAはTLR9,細胞質内に遊離した自己DNAはcGAS,自己RNAはTLR3やRLRによって認識される.ATPや尿酸結晶などのDAMPsはNLRP3インフラマソームの活性化を誘導する.また,組織損傷時に生じる細胞外マトリックスの構成成分であるバイグリカンやヒアルロン酸はTLR2, TLR4を活性化する.このように,自然免疫細胞はPAMPsだけでなく細胞死や組織損傷によって生じるDAMPsによっても活性化され,さまざまな炎症反応を誘導する.それらの反応は,病原体の排除や組織修復において重要な役割を果たすことや獲得免疫誘導に働く場合がある.抗がん剤や放射線により死滅したがん細胞から放出した自己DNAがcGAS-STING経路を活性化することで抗腫瘍免疫応答に寄与することや,ウイルス感染により死滅した細胞由来のHMGB1がT細胞応答に重要であることが示されている24–26).一方,意図しない炎症反応を誘導することによりさまざまな炎症性疾患の原因ともなりうる23).
生体に備わる核酸分解酵素は,病原体由来の核酸を分解することで病原体の複製を阻害する役割を果たすと同時に,PRRsによる認識を可能とすることで自然免疫応答誘導に貢献する.2′-5′オリゴアデニル酸合成酵素OAS1がウイルスRNAと結合するとセカンドメッセンジャーである2′-5′オリゴアデニル酸が産生され,これによりRNase Lが活性化される.その結果,RNase LがウイルスRNAを分解しウイルス複製を抑制するとともに,分解された内在性RNAがRLRを活性化することで抗ウイルス自然免疫を強化する27).また,RNase T2やRNase 2によりリソソームで分解されたウイルスや細菌RNAは,TLR7やTLR8のリガンドとして機能する28, 29).DNAに関しては,リソソームにおけるCpGを含む病原体DNAのDNase IIによる分解がTLR9活性化に重要である30).しかしながら,DNase II阻害は,長鎖の自己DNAを生じさせ,これがTLR9経路活性化を誘導し自己免疫疾患につながることが示唆されている31).また,DNase III/TREX1による細胞質内DNAの分解はcGAS-STING経路による認識を回避するために重要である31).また,PRRsやその下流シグナル因子の変異による恒常的な活性化がさまざまな自己免疫疾患や自己炎症性疾患で認められている31).Singleton–Merten症候群(SMS)の疾患の一部では,RIG-IやMDA5のATP結合部位に変異が生じており,それによる過剰なI型IFN産生が病態に関わっている.また,MDA5の変異は全身性エリテマトーデスやAicardi–Goutières症候群の患者でも認められている.肺や皮膚の炎症を示すSAVI症候群ではSTINGの恒常的活性型変異が認められ,COP-I小胞αサブユニットに変異が生じるCOPA異常症ではSTINGの細胞内輸送の異常による恒常的な活性化が認められる32, 33).一方,自己核酸が内在性のタンパク質と結合することでPRRsアゴニストへと質が変化し,疾患と関連することも報告されている34).乾癬においては,ケラチノサイトから産生される抗菌ペプチドLL-37と自己DNAとの複合体が形質細胞様樹状細胞上のTLR9を活性化することで炎症が悪化することが示されている.乾癬モデルマウスを用いた研究では,TLR9欠損により病態が改善する.
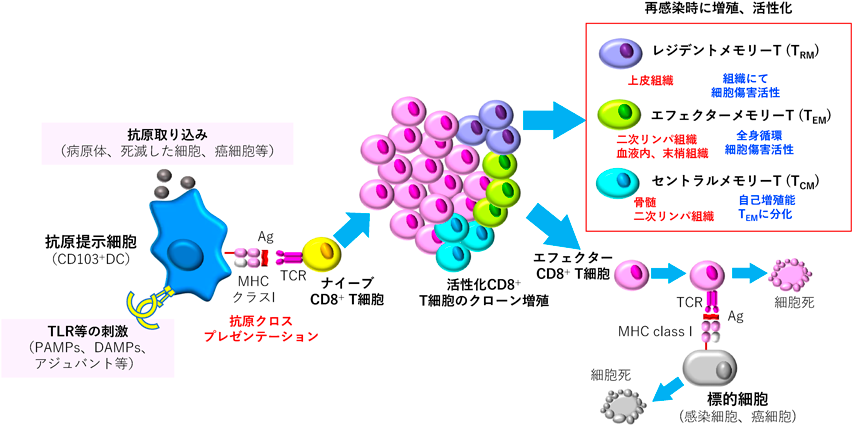
獲得免疫系の成立には,マクロファージや樹状細胞といった抗原提示細胞によるMHC分子への抗原提示に加え,PRRsを介したPAMPs認識によるサイトカイン類の産生や共刺激分子の細胞表面発現が必要である(図1).抗原提示細胞の中でも,樹状細胞は感染局所で病原体を取り込むと,リンパ節へと移動しナイーブT細胞と相互作用する.MHC分子にはMHCクラスIとMHCクラスIIが存在するが,抗原提示の経路は異なる.MHCクラスIIはマクロファージや樹状細胞といった貪食能を有する細胞に発現している.これら細胞に取り込まれた抗原はエンドソーム内で分解され,生じた抗原ペプチドはMHCクラスII分子と結合し提示される.この複合体を認識したナイーブCD4陽性T細胞は,エフェクターヘルパーT細胞へと分化し,B細胞やキラーT細胞,マクロファージ等の活性化を補助する.この経路は,外来性抗原の貪食により行われる.一方,細胞内に感染したウイルス由来の抗原やがん細胞のもつ変異タンパク質に由来する抗原は細胞内のプロテアソームで分解され,小胞体内でMHCクラスI分子と抗原ペプチドが結合し提示される.この経路により内在性抗原が提示される.MHCクラスI分子–抗原ペプチド複合体はナイーブCD8陽性T細胞に抗原提示されエフェクターキラーT細胞へと分化する.このクラスI拘束性経路は抗原提示細胞のみならずほぼすべての細胞に存在する.一方,外来性抗原をMHCクラスIに提示するクロスプレゼンテーションという機構も存在する(図4)35).これは,特定の樹状細胞サブセットが持つ仕組みであり,外来病原体や,死滅した感染細胞を細胞内に取り込んだ後,分解した抗原ペプチドをMHCクラスI分子上に提示する.この仕組みにより,クラスIの発現を抑制する仕組みを持つウイルスや抗原提示細胞に感染しないウイルスに対して適切なキラーT細胞応答(細胞性免疫)が誘導されると考えられる.
エフェクターT細胞の多くは抗原接触後1~2週間で死滅するが,一部はメモリーT細胞として体内で長期にわたり生存する(図4).CD8陽性メモリーT細胞は大きくエフェクターメモリーT細胞(TEM),セントラルメモリーT細胞(TCM),組織常在型(レジデント)メモリーT細胞(tissue-resident memory T cell:TRM)の三つに分けられる36).TEMは脾臓やリンパ節等の二次リンパ器官や血液内,末梢組織に幅広く存在する.TCMは骨髄や二次リンパ器官に多く存在する.一方,TRMは主に肺や腸などといった上皮組織に常在しており,組織間移動を行わないとされている.TEMは再度病原体に感染したときに速やかに反応し,細胞傷害活性を示すと同時にエフェクターCD8陽性T細胞にも分化する.TCMは自己増殖能を有しており,メモリーCD8陽性T細胞の維持に寄与しているのみならず,TEMに分化することも知られている.TRMはナイーブT細胞およびTCMから分化したTRM前駆細胞から分化し,上皮組織や腫瘍組織において細胞傷害活性を示すと考えられている.
このように細胞性免疫は,感染細胞やがん細胞の排除に中心的な役割を果たすが,現在,主に使用されている不活化ワクチンは抗体による体液性免疫を誘導するものの,細胞性免疫の誘導は十分でないと考えられている.また,長期免疫記憶を誘導することも難しい.弱毒生ワクチンでは体液性免疫と細胞性免疫の誘導と長期免疫記憶が誘導可能であるが,変異株の出現や感染リスクもゼロではない.また,現在使用されているアジュバントであるアルミニウム塩(アラム)は2型ヘルパーT細胞の活性化を通して体液性免疫を主に誘導する.細胞性免疫の誘導には,mRNAワクチンやDNAワクチン,ウイルスベクター等の核酸を用いた内在性抗原の発現,クロスプレゼンテーションを強化するアジュバント,クロスプレゼンテーションを行う細胞の利用等の工夫が必要である.次節では,肺組織特異的にTRMを誘導する仕組みについて我々の最近の知見を紹介する.
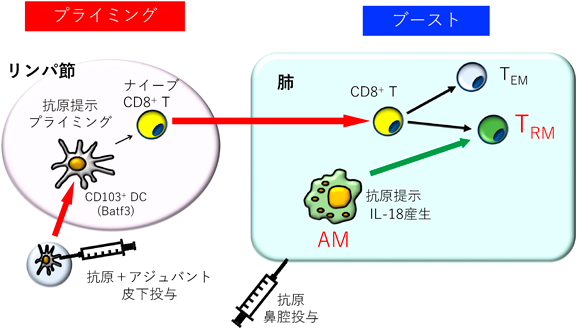
5. 抗原鼻腔投与による肺内でのキラーT細胞の誘導
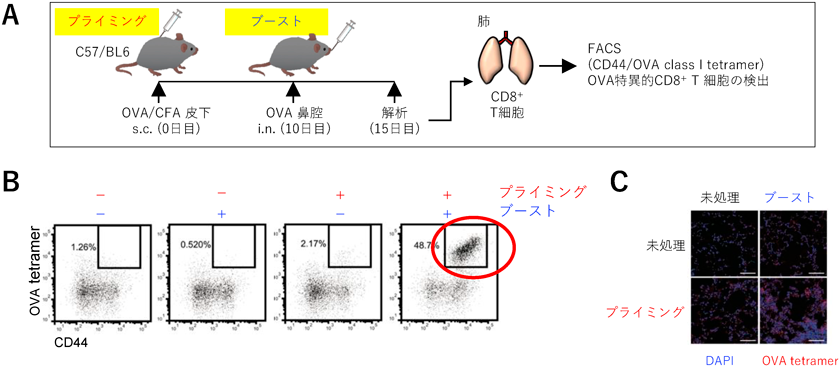
ワクチン投与ルートや使用するアジュバントにより免疫応答は異なることが知られている.そこで,皮下,腹腔,静脈,鼻腔投与等の投与ルートやさまざまなアジュバントを用いて検討を行った.マウスに完全フロイントアジュバントとともにモデル抗原である卵白アルブミン(OVA)を皮下注射(プライミング)し,10日後に抗原のみ鼻腔投与(ブースト)を行い,その5日後に解析したところ肺内でOVA特異的CD8陽性T細胞が誘導されることを見いだした(図5)37).また,これら細胞は活発に増殖していた.FTY720投与によりリンパ節からのT細胞の移動を阻害した状態においても肺内でのOVA特異的CD8陽性T細胞の増殖は阻害されなかったことから,肺組織に常在しているCD8陽性T細胞が抗原鼻腔投与後に増殖したものと考えられた.完全フロイントアジュバントは肉腫形成等の点からヒトでは用いることができないため,ヒトで使用されているアジュバントAS04(TLR4アゴニストであるモノホスホリピドAを含む)を用いて試したところ,同様の結果を得ることができた.一方,別のアジュバントであるアラムでは,完全フロイントアジュバントと同程度の抗体産生を誘導したものの肺におけるCD8陽性T細胞の誘導は認められなかった.このように,アジュバントにより最終的な獲得免疫応答は異なるが,その分子機構は不明な点が多い.
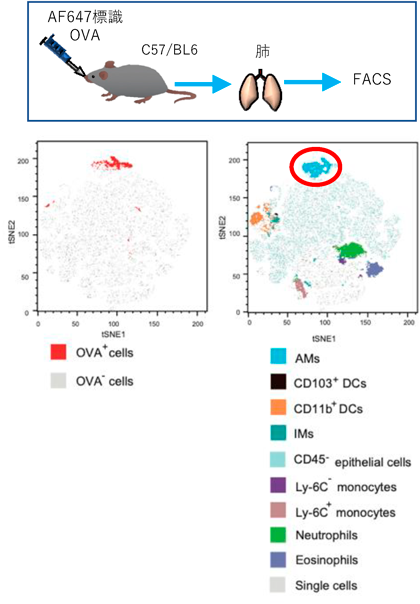
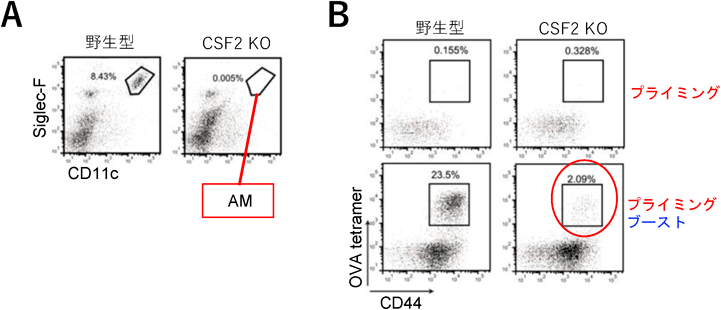
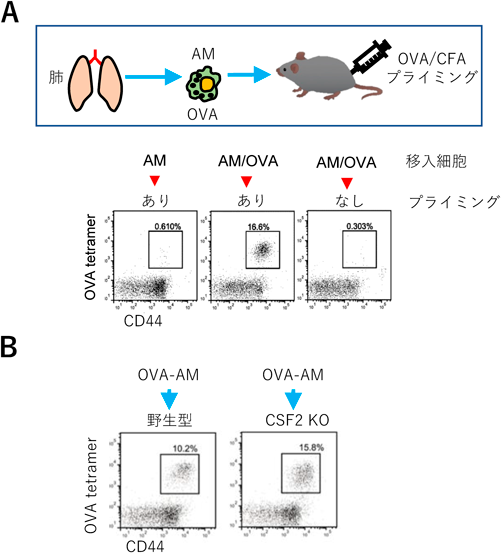
マウス肺内には肺胞マクロファージ(AM),間質マクロファージ,CD103陽性樹状細胞,CD11b陽性樹状細胞,単球,好中球,好酸球等さまざまな種類の自然免疫細胞が局在しており,感染防御や恒常性維持に貢献している38).そこで,抗原鼻腔投与後のCD8陽性T細胞増殖に寄与する自然免疫細胞を解析したところ,AMが優先的に抗原を取り込んでいた(図6).AMは,肺胞に特異的に存在する組織常在型マクロファージであり,肺胞腔内に侵入した病原体や微粒子の貪食と排除を行うことで感染防御やアレルギー性炎症の抑制などに寄与している.また,サーファクタントや死滅した上皮細胞の取り込み分解を行うことで肺胞内の恒常性維持に寄与している.その分化・成熟には主に顆粒球マクロファージコロニー刺激因子(granulocyte-macrophage colony-stimulating factor:GM-CSF)が関与しており,GM-CSFまたはGM-CSF受容体欠損マウスでは,AMの枯渇を伴う肺胞タンパク質症を発症する.ヒトにおいてもGM-CSFに対する自己抗体を持つ自己免疫性肺胞タンパク質症が知られている.AMの分化・成熟にはPPARγ, Bach2, C/EBPβといった転写因子や我々が見いだした脂質リン酸化酵素PIKfyveが重要な役割を果たしている38–40).一方,AM自身が抗原提示細胞として機能しているかは不明な点が多い.そこで,OVAを貪食させたAMとOVA特異的TCRトランスジェニックマウス(OT-Iマウス)由来のCD8陽性T細胞をin vitroで共培養したところT細胞の増殖が認められた.一方,MHCクラスIへの抗原提示に必須の役割を果たすトランスポーターTAP-1を欠損したマウスのAMを用いた場合,CD8陽性T細胞増殖が誘導されなかったことから,AMはMHCクラスIへの抗原提示依存的にCD8陽性T細胞増殖を誘導することがわかった.次に,AMを持たないマウスを用いて検討を行った.クロドロネートリポソームを用いてAMをマウス体内より除去したところ,ブースト後の肺内CD8陽性T細胞の増殖が阻害されていた.さらに,GM-CSFノックアウトマウス(CSF2 KO)を用いて解析したところ,同様に肺内CD8陽性T細胞の誘導が阻害されていた(図7).また,OT-I CD8陽性T細胞を野生型マウスに移入すると,ブースト後に肺内CD8陽性T細胞の増殖が認められたが,GM-CSF欠損マウスに移入した場合には認められなかった.一方で,プライミングしたGM-CSF欠損マウスにOVAを取り込ませたAMを移入するとブーストなしで肺内CD8陽性T細胞の増殖が誘導された(図8).この結果は,プライミング後に誘導されるリンパ節内でのナイーブCD8陽性T細胞の活性化とエフェクター細胞への分化にAMは必須ではないことも示唆する.クロスプレゼンテーションにおいて中心的な役割を果たす樹状細胞サブセットとして知られているCD103陽性樹状細胞の分化は転写因子Batf3により制御されている.そこで,Batf3欠損マウスを作製し,解析を行った結果,このマウスではプライミングとブースト後の抗原特異的CD8陽性T細胞の肺内での誘導が欠如していた.さらに,OVAを取り込ませた野生型AMをプライミング済みのBatf3欠損マウスに移植してもCD8陽性T細胞の誘導は認められなかった.このことは,AM欠損マウスとは異なり,CD103陽性樹状細胞を持たないマウスでは,プライミングが誘導されていないことを強く示唆するものである.したがって,CD103陽性樹状細胞は主にプライミング時におけるナイーブCD8陽性T細胞の活性化に重要な役割を果たす一方,AMはプライミングに寄与せずブースト後のCD8陽性T細胞の増殖に寄与するものと考えられる.
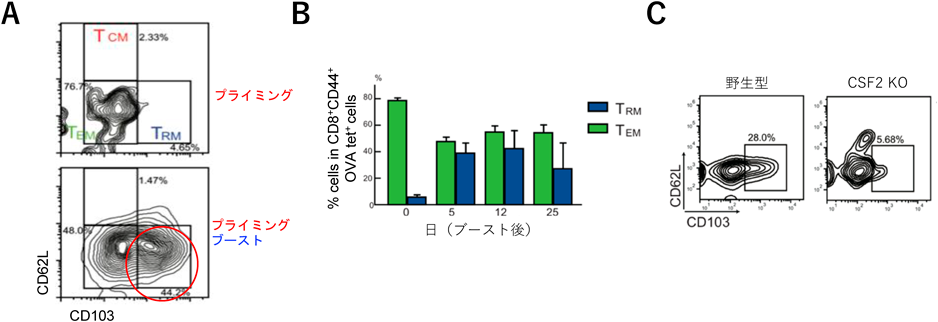
次に,この免疫法により肺内で誘導された抗原特異的CD8陽性T細胞の詳細を解析したところ,CD103陽性CD62L陰性TRMの割合が有意に増えていることがわかった(図9).また,CD8陽性TRMはブースト後25日時点でも維持されていたことから,メモリー細胞であることが示唆された.GM-CSFノックアウトマウスで解析したところ,CD8陽性TRMは野生型マウスと比べ顕著に減少していた.そこで,AMによる肺内CD8陽性TRMの増加を制御するメカニズムについて解析を行った.1細胞解析のデータセットを利用し,AMで発現の高い遺伝子を抽出したところ,IL-18を見いだした.IL-18は主にマクロファージ等の自然免疫細胞から感染刺激等により分泌される.その機能は多岐にわたっているが,IL-12の存在下でCD4陽性T細胞に作用しIFNγ分泌を促すことでTh1型細胞応答を誘導することが知られている.そこで,IL-18受容体ノックアウトマウスを用いて解析を行ったところ,ブースト後の抗原特異的CD8陽性TRMの誘導が野生型マウスと比べ有為に減少していた.この背景に,IL-18の作用による1型ヘルパーT細胞(Th1細胞)からのIFNγ等の分泌が肺内CD8陽性TRMの増加に関与している可能性が考えられた.そこで,MHCクラスIとクラスIIで提示されるいずれの抗原を含むOVAタンパク質ではなく,MHCクラスIおよびクラスII拘束性のペプチドを用いてブーストを行い,Th1型細胞の関与を検討した.その結果,MHCクラスI拘束性ペプチドのみでブーストしたときと比べ,クラスI拘束性ペプチドとクラスII拘束性ペプチドで同時にブーストした場合に肺内CD8陽性TRMの顕著な増加が認められた.一方,IL-18受容体欠損マウスでは,この増加が減弱していた.したがって,抗原ブーストに伴いAMから産生されるIL-18によりTh1型細胞からのIFNγ産生が誘導され,これによりCD8陽性TRMの増殖が強化されたものと考えられた.
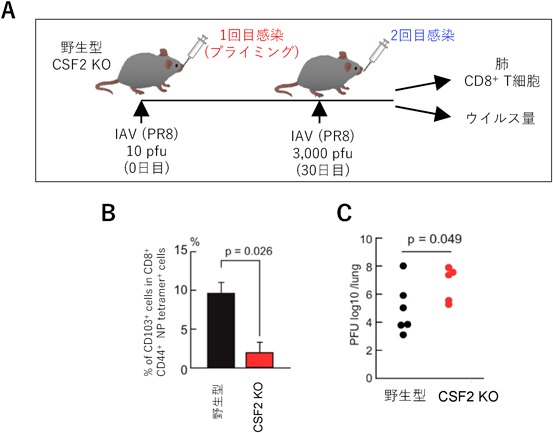
最後に実際のウイルス感染後のCD8陽性T細胞応答にAMが関与しているか検討を行った.マウスに少量(10 pfu)のインフルエンザA型ウイルスPR8株を鼻腔から感染(プライミング)させ,30日後に大量(3000 pfu)の同ウイルスを鼻腔より再感染(ブースト)させ,5日後に解析を行った.プライミングしたマウスではブースト後にウイルス特異的CD8陽性TRMが肺内で顕著に増加していた.また,プライミングは大量感染による肺の炎症を抑制した.GM-CSF欠損マウスの肺では,ブースト後5日目でのウイルス特異的CD8陽性TRMが減少しておりウイルス量が増加していた(図10).さらに,プライミングしたGM-CSF欠損マウスに野生型AMを移植後,ブーストを行うと肺内のウイルス特異的CD8陽性TRMが野生型マウスと同様に誘導された.以上のことから,AMはインフルエンザウイルス再感染時におけるメモリーCD8陽性T細胞の増殖と肺での感染防御に重要な役割を果たしていることが示唆された.
今回の結果から,鼻腔からの抗原のブーストやウイルス再感染時における肺内でのTRMの急激な増殖にAMが中心的な役割を果たしていることが明らかとなった(図11).また,この増殖には,AMによる抗原クロスプレゼンテーションとIL-18産生の両方が重要であった.一般的に,ワクチン接種において,抗原をアジュバントとともに皮下や筋肉内に複数回行うことで高い抗体価や細胞性免疫の誘導と免疫記憶が誘導される.一方で,アジュバントは自然免疫系を活性化することで効率よく獲得免疫を誘導するが,炎症等の副作用の懸念も生じる.今回我々の用いた方法は,ブースト時にアジュバントを必要としないにもかかわらず高い免疫記憶と細胞性免疫を肺局所で誘導することができる特徴を持つ.そのため,安全性という点において本手法は有用であると期待される.肺に常在するCD8陽性TRMは肺組織における感染細胞や腫瘍細胞の排除に寄与することから,新型コロナウイルスを含む気道感染型ウイルスにより引き起こされる重症化や腫瘍の肺転移等を抑制する可能性が考えられる.本手法の汎用性は高いことも期待される.興味深いことに,本研究ではin vitroで抗原を貪食させたAMをプライミング済みのマウスに移植することでブースト無しでも抗原特異的CD8陽性TRMが誘導されたことから,AMが細胞移植型ワクチンとして利用できることが期待される.たとえば,iPS細胞や単球から分化させたAMを用いた自家移植は,インフルエンザウイルスや新型コロナウイルスのみならず肺腫瘍に対するCD8陽性TRMの誘導に有用な手法となると期待される.特に,肺障害や老化等によりAMの数や機能が低下しており十分なTRMが誘導できない状況において有用であると期待される.
肺内CD8陽性TRMは,新型コロナウイルスによる重症化防止に大きく寄与すると考えられるものの,現在使用されているmRNAワクチンでは肺局所での長期免疫記憶や細胞性免疫が十分に獲得されるとは言いがたい.最近,エール大学の岩崎らのグループは,肺局所においてこれらを誘導する免疫手法を確立し報告した41).SARS-CoV2スパイクタンパク質をコードするmRNAワクチンをマウスの筋肉内に投与しプライミングした後,アジュバントを含まない抗原タンパク質を鼻腔投与することでブーストを行ったところ,抗原特異的なメモリーT細胞やメモリーB細胞が肺内に誘導された.また,この免疫によりマウスやハムスターにおけるウイルス感染後の重症化を抑制した.彼女らは,この免疫手法をprime & spritと名づけ,肺局所におけるメモリー細胞の誘導が長期免疫記憶の確立や重症化防止に重要であると提唱している.さらに,T細胞は変異した抗原に対してヘテロに対応することができることから,prime & spritは変異に対しても対応可能な手法であることも示している.今後,我々の手法も含め,肺局所で獲得免疫を誘導することで感染防止や重症化予防に貢献するワクチンの開発が進むことが期待される.