翻訳後修飾は,タンパク質の細胞内局在や安定性,酵素活性などを変化させて,その機能を制御する機構であり,これまでに200種類以上が同定されている3).このうち,リン酸化がERK経路の活性制御に重要な役割を果たすことはよく知られているが,近年,リン酸化以外の翻訳後修飾による調節機構も多数見いだされている.さらにERK経路の下流で発現誘導される分子を介したフィードバック制御などの存在も明らかにされており,その多彩な制御システムの全体像が明らかにされつつある(図2).
1)リン酸化・脱リン酸化によるERK活性の制御
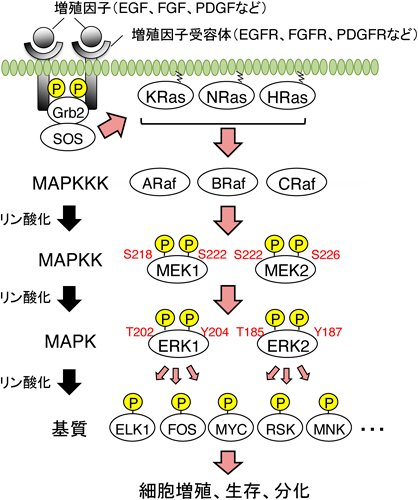
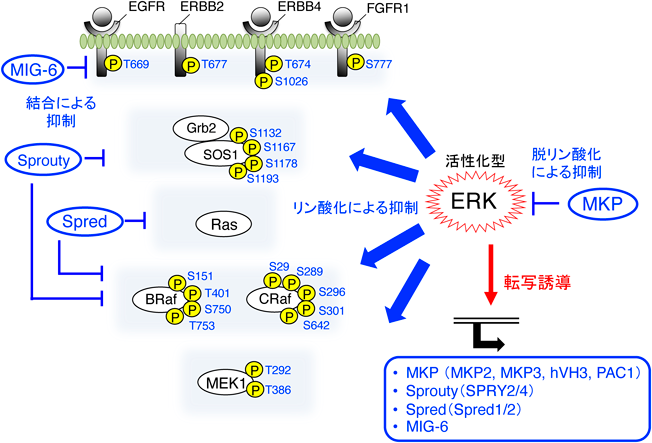
リン酸化は生物における普遍的な翻訳後修飾の一つであり,リン酸化酵素(キナーゼ)と脱リン酸化酵素(ホスファターゼ)のバランスによって調節されている.増殖因子受容体からERKの活性化に至る機構に関しては,すでに多くの優れた成書が存在することから本稿ではふれないが,ERKカスケード(Raf–MEK–ERK)においては,各キナーゼ分子間の連続したリン酸化反応によってシグナルが伝播されている.特にERKは,酵素ドメインの活性化ループ内に存在するトレオニンおよびチロシン残基が,MEKによってリン酸化されて活性化することから,ホスファターゼによる同残基の脱リン酸化はERK活性を阻害することになる.これまでにERKを選択的に脱リン酸化するMAPK phosphatase(MKP)として,6種類の分子[PAC1(DUSP2),MKP2(DUSP4),hVH3(DUSP5),MKP3(DUSP6),MKPX(DUSP7),MKP4(DUSP9)]が同定されている4, 5)(図2).各MKP分子は,それぞれ異なる細胞内局在,活性制御機構,および組織特異性を示すことが知られており,MKP3, MKPX, MKP4が主として細胞質に局在するのに対し,PAC1, MKP2, hVH3は核内に優位に局在することが明らかにされている.これらの分子のうち,MKP2, MKP3, PAC1, hVH3はERKの活性化に応答して転写誘導される分子であり,ERKを脱リン酸化して抑制することで,その活性持続時間を制限するネガティブ・フィードバックループを形成している6).これらERK誘導型MKPの転写制御機構に関しては,最近,ERKの下流で活性化するribosomal S6 kinase(RSK)が,転写抑制因子capicua transcriptional repressor(CIC)のS173/S301残基をリン酸化することで14-3-3との結合を促し,CICの核外排出を導くこと,またその結果,CICによるMKPの転写抑制が解除されその発現が増強することが示されている7).さらに,タンパク質レベルでの発現調節機構も報告されており,K48結合型ポリユビキチン化(MKP2, MKP3, hVH3, MKPX)や,ERK依存的リン酸化(MKP3, MKP2)によってMKPタンパク質の分解が促進されることも見いだされている8).また,DUSP8/hVH5はこれまでp38とJNKに選択的なMKPと考えられてきたが,近年,ERKの制御因子としても機能することが示されている9).DUSP8は脳,心臓,筋肉などで強く発現しているが,実際にDUSP8遺伝子欠損マウスでは,脳,心臓におけるERK活性の増強と心筋肥大が観察される10).心臓特異的にERKを活性化させたトランスジェニックマウスでも,同様の心筋肥大が認められることから11),少なくとも心臓においては,DUSP8が過剰なERK活性を抑制することで,正常な臓器発生に寄与していると考えられる.
ERKシグナルを負に制御する機構としては,上述のMKPによる脱リン酸化のみならず,ERKが自身の上流に位置する分子を直接リン酸化してその機能を阻害するという,ネガティブ・フィードバック機構の存在も明らかにされている12, 13)(図2).たとえば,活性化したERKは,SOS114)や,BRaf, CRaf15, 16),MEK117, 18)などをリン酸化し,各分子の機能を抑制する.またEGFRやERBB2, ERBB4などの受容体型チロシンキナーゼもERKによってリン酸化されるとキナーゼ活性が低下する19, 20).このようなERKによる上流因子のフィードバック・リン酸化は,ERKシグナルの活性持続時間と強度を規定し,後に続くさまざまな生命現象(増殖や分化など)の厳密な制御に重要な役割を果たしている.さらに,ERK以外のキナーゼによる制御も報告されており,protein kinase C(PKC)やcGMP-dependent protein kinase G(PKG)が,KRas(S181)をリン酸化して膜局在を阻害し,ERK経路を抑制することが示されている21, 22).また反対に,細胞が細胞外マトリックスへ接着した際に活性化するp21-activated kinase 1(PAK1)は,MEK1(S298)をリン酸化することで,MEK1とERK間の結合親和性を高め,足場依存性増殖を促すことが明らかにされている23).
2)リン酸化以外の翻訳後修飾による活性制御機構
近年,ERK経路構成因子の機能制御に,リン酸化以外にも多彩な翻訳後修飾が関与することが明らかにされている(表1).Rasは翻訳後,直ちにC末端がファルネシル化されて脂質二重膜と結合できるようになり,これにより生理的な活性化の場である細胞膜に局在する.この他にも,ADP-リボシル化,ニトロシル化,アセチル化,ユビキチン化などの翻訳後修飾がRasの活性を正・負に制御することが報告されている24).またRafも複数の修飾を介してその発現量や酵素活性が調節されている.たとえば,BRafとCRafはK48結合型ポリユビキチン化によりプロテアソーム依存的に分解されることが示されており,このユビキチン化を担うE3リガーゼとして,BRafではRNF149, FBXW7, APCFZR1が,一方,CRafでは,CHIP, HUWE1, HERC1, CTLHが同定されている25–29).また,反対にRafを脱ユビキチン化し,安定化する酵素としてUSP10, USP13, USP15が同定されている30–32).さらに最近,分解系のユビキチン修飾のみならず,HECTタイプのE3であるITCHがBRafにK27結合型ポリユビキチン鎖を付加することも報告された33).K27結合型ポリユビキチン鎖は,PP2A型ホスファターゼが結合する足場となって,BRafの抑制的リン酸化サイトであるS365の脱リン酸化を促すことでBRaf活性を遷延化させることが示されている.また最近,ヒストンアセチル化酵素p300が,BRaf(K601)をアセチル化して,BRaf分子どうしのホモ二量体化や,BRafとCRafもしくは足場タンパク質KSR1とのヘテロ二量体化を促進し,ERKシグナルを増強することも報告されている34).
表1 リン酸化以外の翻訳後修飾によるERKシグナル構成因子の活性制御 | 標的タンパク質 | 修飾 | 機能 | 修飾酵素 |
|---|
| 受容体 | EGFR | mono-/poly-Ub (K48/K63) | エンドサイトーシス/分解 | c-Cbl, Cbl-b, CGRRF1, ZNRF1, HUWE1, RNF126, SMURF2 |
| PDGFR | mono-/poly-Ub | エンドサイトーシス/分解 | c-Cbl, Cbl-b, TRIM21 |
| TGFβR (TβRI) | SUMO1 | Smad3結合・リン酸化促進 | unknown |
| アダプター分子 | Grb2 | SUMO1 | SOSとの結合増強 | unknown |
| Ras | HRas, NRas | mono-/di-Ub | エンドサイトーシス | RabGEF1 |
| KRas, HRas, NRas | mono-/di-Ub | GTP結合型維持 | unknown |
| poly-Ub | タンパク質分解 | β-TrCP |
| ニトロシル化 | GDP/GTP交換促進 | eNOS, iNOS, nNOS |
| KRas | SUMO3 | Raf結合亢進 | PIASγ |
| アセチル化 | GDP/GTP交換阻害 | p300 |
| MAPKKK | BRaf | poly-Ub | タンパク質分解 | RNF149, FBXW7, APCFZR1 |
| poly-Ub (K27) | キナーゼ活性亢進 | ITCH |
| アセチル化 | キナーゼ活性亢進 | p300 |
| CRaf | poly-Ub | タンパク質分解 | CHIP, HUWE1, HERC1, CTLH |
| MAPKK | MEK1 | SUMO1 | キナーゼ活性阻害 | MEKK1 (SUMO-E3) |
| アセチル化 | キナーゼ活性阻害 | p300 |
| 切断 | キナーゼ活性阻害 | Caspase-3 |
| MEK2 | SUMO1 | キナーゼ活性阻害 | MEKK1 |
| MAPK | ERK1/2 | poly-Ub | タンパク質分解 | MEKK1 |
| ISG15 | unknown | unknown |
| ERK1c | mono-Ub | ゴルジ体断片化 | unknown |
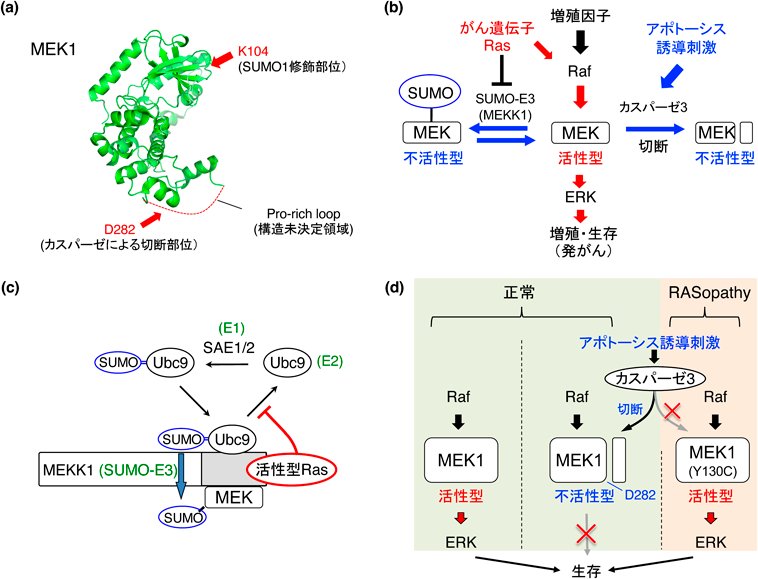
Rafと同様,MEKもさまざまな翻訳後修飾を介してその活性が厳密に制御されている.筆者らは,MEKがsmall ubiquitin-like modifier 1(SUMO1)化されること,またその結果,基質であるERKとの結合が阻害されて,ERK活性が負に制御されていることを報告した35)(図3a, b).実際にSUMO化不能型のMEK1(K104R)変異体を発現する細胞では,増殖因子刺激によるERK経路の活性化が増強して増殖能が有意に亢進したことから,MEKのSUMO化は,ERKの過剰な活性化を防ぎ,増殖シグナルを適切なレベルに制御する上で重要な役割を果たしていると考えられる.さらに我々は,がん遺伝子である活性型Rasが,MEKのSUMO化を阻害する作用を持つことを発見し,実際にRasに変異を有するさまざまなヒトがん細胞においてMEKのSUMO化が消失していることを確認した.また反対に,MEKのSUMO化を強制的に亢進させると,活性型Rasによる細胞の悪性形質転換が有意に抑制されることも確認した.すなわち,がん遺伝子Rasは,Rafを活性化すると同時に,MEKのSUMO化修飾による不活性化を阻止する,という二重の機構によってERK経路を強くそして効率よく活性化し,発がんを導いていることを示した(図3b).
次に我々は,活性型RasによるMEK-SUMO化の阻害機構を解明するため,MEKのSUMO化を制御する分子の同定を試みた.その結果,MAPKKKの一種であるMEKK1が,MEKのSUMO化を担うE3リガーゼとしても機能していることを見いだした(図3c).さらに,MEKK1とRasとの相互作用を解析したところ,がん遺伝子RasがMEKK1と直接結合して,MEKK1(E3)とUbc9(E2)の結合を著しく増強させることを突き止めた.E1, E2,およびE3分子は,相互に結合と解離のサイクルを繰り返すことで,標的タンパク質を構成的にSUMO化することが知られている.活性型Rasは,MEKK1とUbc9の結合を増強し,解離を阻害することで,このSUMO化のサイクルを停止させ,MEKのSUMO化を抑制していると考えられる.以上の結果から,がん遺伝子Rasは,MEKK1のSUMO-E3リガーゼ活性を阻害し,MEKのSUMO化を阻止するというユニークな機能を持つことが明らかとなった.
SUMO化によるERK経路の制御としては,近年,我々の知見以外にもいくつかの例が報告されている.たとえばKRasは,E3リガーゼであるPIASγを介してSUMO3化されると,Rafへの結合能が増強する36, 37).実際にSUMO化不能型KRas(K42R)変異体を発現する細胞では,遊走能や浸潤能が低下することから,SUMO3修飾はKRas活性を補強する機構であると考えられる.またGrb2(K56)のSUMO1修飾は,SOSへの結合親和性を増大させてRasの活性化を促進する38).さらに,細胞内タンパク質のグローバルなSUMO化レベルの亢進が,がんの進行や予後と相関することも示されており,実際に膵・肺・大腸がんなど,多くのがん組織でSUMO化関連酵素(E1:SAE1-SAE2, E2:Ubc9)の過剰発現や,脱SUMO化酵素(SENP)の減少が認められる39).現在,これらSUMO化修飾酵素を標的とした抗がん剤の開発も進められており,特にE1阻害剤TAK-981はリンパ腫および転移性固形腫瘍を対象とした臨床試験が進行中である40).
また我々は最近,アポトーシスの実行酵素であるカスパーゼ3が,MEK1の酵素ドメイン内に存在するVEGD282配列を直接切断してキナーゼ活性を不可逆的に阻害し,ERK経路を遮断することを見いだした41)(図3a, b, d).ERKはさまざまなアポトーシス促進分子(FOXO3a, Bim,カスパーゼ8/9など)をリン酸化してその機能を阻害し,細胞死を抑制することが知られている.したがって,カスパーゼ3によるMEK1の切断は,生存シグナルであるERK経路を不活性化して,アポトーシスをさらに亢進させる機構であると考えられる.実際に,切断不能型MEK1(D282N)変異体を発現する細胞では,DNA損傷や高浸透圧などのストレス環境下でもERK活性が高いまま維持され,これらのストレスによるアポトーシス誘導が有意に抑制された.また興味深いことに我々は,後述する先天性RASopathy患者で高頻度に認められるMEK1(Y130C)変異によって,カスパーゼ依存的なMEK1の切断が完全に消失することも発見した(図3d).これらの結果から,カスパーゼ3依存的なMEK1の切断によるERK経路の制御は,人体の正常な発生に寄与しており,その破綻がRASopathyの病態形成にも寄与していると考えられる41).
3)ERKシグナル誘導遺伝子による活性制御
ERK経路の活性化に伴って発現誘導され,同経路を負に制御する遺伝子として,上述したMKP以外にも複数の分子が同定されている.Sprouty(SPRY)およびSpredは,ショウジョウバエから哺乳類に至る広汎な真核生物において保存された分子であり,ともにERK経路依存的に転写誘導される.ERKの活性化に伴って発現したSPRYはGrb2と結合して,Grb2-SOS複合体の形成を競合的に阻害し,Rasの活性化を抑制する.またSPRYはRafとも結合して,そのキナーゼ活性を抑制することが報告されている42)(図2).SpredはRas-GAP(GTPase-activating protein)であるNF1と結合して細胞膜へリクルートすることでRasの活性化を阻害する.SPRYやSpredの発現は,大腸・肝・乳がんなど,さまざまながんで低下していることから,がん抑制遺伝子として機能すると考えられる43).また,MIG-6(mitogen-inducible gene-6)もERK経路依存的に発現誘導され,EGFRと相互作用してその自己リン酸化を阻止することで不活化する44).このようにERKは,自身の上流に位置する分子(SOS, Raf, MEKなど)を直接リン酸化してその機能を抑制する機構と,負の制御因子(MKP, SPRY, Spread, MIG-6など)の発現誘導を介して間接的に活性阻害を導く機構という,2種類のネガティブ・フィードバック機構を使い分け,活性持続時間と強度を厳密に制御している.
ERKはプロリン指向性キナーゼであり,基質分子内に存在するS/T-P配列内のセリンまたはトレオニン残基をリン酸化する.ERKの基質特異性は,このリン酸化サイトの配列選択性に加えて,ERK-基質分子間のタンパク質間相互作用(docking interaction)によっても規定されている.基質分子や,MEK, MKPなど,ERKと選択的に相互作用する分子は,その内部にDサイトと呼ばれる特徴的なアミノ酸配列が存在することが知られており,このDサイトを介してERKのC末端に存在するCDサイト(common docking site)と結合する.すなわち,ERKのCDサイトが基質分子内に存在するDサイトを選択的に認識して結合することで,ERKの基質特異性が担保されている45).また,一部の基質分子は,DEFサイト(docking site for ERK FXF)と呼ばれるドッキングサイトを有しており,このDEFサイトを介してERKと結合し,選択的にリン酸化されることも明らかにされている46)
ERKの基質としては,これまでに転写因子やキナーゼ,細胞骨格系分子など100種類以上が同定されている2, 47).たとえば,活性化したERKはその一部が核内へ移行し,Elk1やSP1など複数の転写因子をリン酸化する.ERKによるリン酸化はこれらの転写因子の転写活性を亢進させ,細胞増殖に必要な初期応答遺伝子(immediate early gene:IEG)の転写を導く48).またERKは,キナーゼ(RSK, MNK)やアポトーシス関連タンパク質(Bim,カスパーゼ8/9など)などもリン酸化してこれらの分子の機能を正負に制御し,タンパク質合成促進や生存に寄与することも報告されている49, 50).しかしながら,いまだ同定されていない基質分子も数多く存在すると考えられており,その解明は生命現象や発がん機構を理解する上でも重要であると思われる.筆者らは最近,ERKの未知基質分子を同定する新たな実験法(酵母3-hybrid法)を開発して,ヒトcDNA発現ライブラリーのスクリーニングを行い,新規分子を複数同定することに成功している.以下,ERK基質分子に関する我々の知見を概説する.
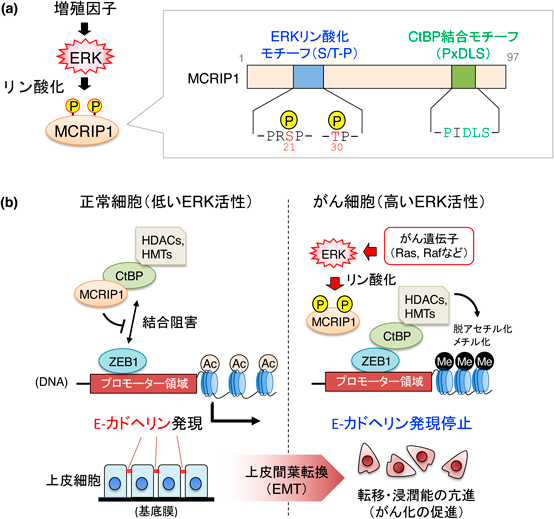
1)MAPK-regulated co-repressor interacting protein 1(MCRIP1)
MCRIP1は,上述のスクリーニングによって単離された約11 kDaの新規ERK基質分子であり,これまでに一切報告がなかったことから我々が命名した遺伝子である51).MCRIP1分子内に,既知の機能ドメインは存在しなかったが,唯一,転写抑制共役因子CtBPと相互作用する特徴的アミノ酸配列(PxDLSモチーフ)が認められ,この配列を介してCtBPと特異的に結合することがわかった(図4a, b).さらにその生理機能について詳細な解析を進めた結果,MCRIP1がCtBPと結合することで,CtBPとZEB1(転写抑制因子)間の結合が競合的に阻害され,CtBP-ZEB1複合体による転写抑制が解除されていることを見いだした.また,興味深いことに,増殖因子やTGFβ刺激などで活性化したERKがMCRIP1をリン酸化すると,MCRIP1がCtBPから解離すること,またその結果,自由となったCtBPはZEB1と結合できるようになって,標的遺伝子(E-カドヘリンなど)のプロモーター上にリクルートされ,最終的にE-カドヘリンの転写抑制および上皮間葉転換(EMT)が導かれることを明らかにした(図4b).さらに我々は,ヒトがんにおけるMCRIP1のリン酸化状態についても解析を行い,各種がん遺伝子の作用によりERK経路が恒常的に活性化している多くのがん細胞で,MCRIP1が異常にリン酸化されていること,またその結果,MCRIP1がCtBPと結合する能力を失ってEMTが起こりやすい状態,すなわち,がん細胞の浸潤・転移が起きやすい状態になっていることを明らかにした.
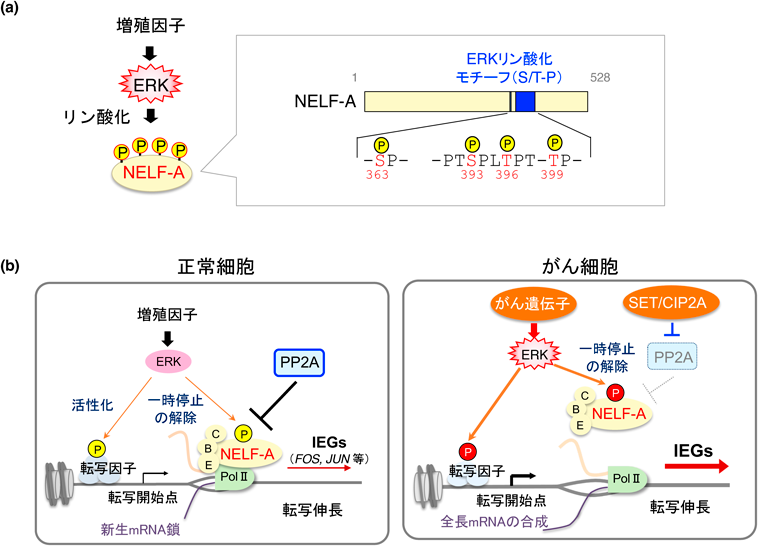
2)negative elongation factor-A(NELF-A)
我々は最近,転写伸長反応制御複合体(NELF)の構成因子であるNELF-AがERKの生理的な基質であることを見いだした52)(図5a, b).ヒトを含む高等生物においては,ゲノムDNA上でRNAポリメラーゼII(Pol II)によるmRNAの転写がスタートすると,直ちにNELF複合体がPol IIと結合して,転写開始点近傍でPol IIを一時停止させ,転写伸張反応を休止させることが知られている.この現象はpromoter-proximal pausing(PPP)と呼ばれているが,無刺激状態の細胞ではPol IIは停止したままとなってmRNAの転写が完結せず,遺伝子発現が低く保たれている.しかしながら,細胞に増殖因子などの外部刺激が加わると,NELF複合体は速やかにPol IIから解離して転写伸長反応が再開し,完全長mRNAが合成されて遺伝子発現が誘導される.このようなNELF複合体を介した転写伸長反応の一時停止とその解除による遺伝子発現制御は,程度の違いはあるもののヒト遺伝子の約60%に認められることが報告されており,特に増殖因子刺激後,迅速かつ同期的に転写誘導される初期応答遺伝子(IEG:JUN, FOS, EGRなど)の発現制御にきわめて重要であることが示されている.しかしながら,増殖因子がどのようにしてNELFをPol IIから解離させ,IEGの転写伸長反応を再開させているのか,そのメカニズムはこれまでほとんど明らかにされていなかった.これに対して我々は,増殖因子によって活性化したERKが,NELF-A分子内の4か所のセリン/トレオニン残基(S363/S393/T396/T399)を直接リン酸化することで,NELF複合体をPol IIから解離させること,またその結果,転写の一時停止が速やかに解除されてIEGの迅速な発現と細胞増殖が導かれていることを明らかにした.加えて,脱リン酸化酵素PP2AがNELF-Aを効率よく脱リン酸化して,IEGの発現を負に制御する分子であることを発見するとともに,ヒトがんではPP2A阻害分子(SETやCIP2A)の異常な高発現によってPP2A活性が低下しており,NELF-Aのリン酸化が亢進してIEGの恒常的な発現とがんの増殖・進展が導かれていることを見いだした(図5b).
1)病原ウイルス・細菌によるERK経路の脱制御
新型コロナウイルスSARS-CoV-2の累計感染者は世界で7億人(2023年3月時点)を超え,現在も増加の一途をたどっているが,その感染過程や病態形成にもERK経路が関与する.これまでにSARS-CoV-2のスパイクタンパク質がPKCを介してERK経路を活性化し,プロスタグランジン合成酵素COX-2の転写を誘導して炎症を悪化させることが報告されている53).また,抗インフルエンザ薬として開発されたMEK阻害剤ATR-002は,少なくともin vitroの細胞培養系において,SARS-CoV-2ウイルスの複製や増殖を阻止するだけでなく,感染細胞から産生されるさまざまな炎症性サイトカイン(IL-6, CXCL8, CCL2など)の発現を抑制することが示されており,COVID-19治療薬としての臨床試験が進められている54).またSARS-CoV-2に限らず,インフルエンザ,エボラ,ヒト免疫不全ウイルスなど,多くの病原ウイルスが,宿主細胞内のERKシグナルを活性化して感染の成立および自身の複製に有利な状況を創り出すことが示されている.特に最近,単純ヘルペスウイルス1型のエンベロープタンパク質gEが,宿主細胞の膜タンパク質prohibitin-1と相互作用してERK経路を活性化すること,またこれによりウイルス粒子の細胞内輸送が亢進して,細胞間のウイルス伝播(cell-to-cell感染)に寄与することが見いだされた55).
ウイルスのみならず病原性細菌も,さまざまなエフェクター分子を産生して宿主細胞に取り込ませ,感染に有利な状況を作り出すが,特に一部の細菌由来分子は,真核生物には存在しない特殊な翻訳後修飾を導く酵素であり,宿主細胞のERK経路を阻害して免疫応答を抑制する機能を持つことが示されている56)(表2).緑膿菌が分泌するExoSは,HRasのR41およびR128をADP-リボシル化して,Rasの活性化に必要なGDP-GTP交換反応を阻害する57).一方,Clostridium sordelliiが分泌するlethal toxinは,HRasのT35をモノグルコシル化し,Ras–Raf間相互作用を破壊してERK経路を抑制する58).サルモネラ菌が分泌するSptPは,チロシンホスファターゼ活性とRasに対するGAP活性を併せ持つ酵素であり,Rafの活性化を阻害する59).また,ペスト菌由来のYopJは,MEKの活性化に必要な2か所のリン酸化サイト(S218/S222)を選択的にアセチル化する機能を有しており,MEKのリン酸化を阻止してそのキナーゼ活性を喪失させる60, 61).炭疽菌が産生するlethal factorは亜鉛結合型メタロプロテアーゼであり,MEKのN末端に存在するERK結合配列(Dサイト)を切断してMEK–ERK間の相互作用を破壊し,シグナル伝達を遮断する62).さらに赤痢菌とサルモネラ菌は,それぞれOspF, SpvCと呼ばれる特殊な酵素(phospho-threonine lyase)を産生して,ERK活性化ループ内のリン酸化トレオニン残基からプロトンを除去するβ脱離反応を引き起こし,デヒドロブチリンへと不可逆的に変換することでERKを不活化する63, 64).このような病原細菌が産生する酵素は,多くの場合ERK経路のみならず,p38/JNK経路やNF-κB経路の構成分子に対しても同様の作用を及ぼし56),宿主細胞の増殖,生存,炎症,免疫応答などを同時に撹乱することで病原性を高めている.COVID-19に対する治療薬としてMEK阻害剤の臨床試験が進められている例が示すように,今後,感染症に対する新たな治療戦略としてシグナル伝達制御薬の活用が期待される.
表2 細菌由来分子によるERKシグナル構成因子の翻訳後修飾| 病原菌 | エフェクター分子 | 宿主内標的タンパク質 | 標的分子への影響 |
|---|
| P. aeruginosa | ExoS | HRas | ADP-リボシル化修飾(R41/R128):GDP/GTP交換阻害 |
| C. sordellii | lethal toxin | HRas | モノグルコシル化(T35):Ras-Raf相互作用阻害 |
| S typhimurium | SptP | CRaf | 活性化阻害(機序不明) |
| Y. pestis | YopJ | MEK1/2 | アセチル化(MEK1:S218/S222, MEK2:S222/S226):キナーゼ活性阻害 |
| B. anthracis | lethal factor | MEK1/2 | N末端切断(MEK1:P8-I9間,MEK2:P10-A11間):キナーゼ活性阻害 |
| S. flexneri | OspF | ERK1/2 | リン酸基脱離化(ERK1:T202, ERK2:T185):キナーゼ活性阻害 |
| S. enterica | SpvC | ERK1/2 | |
2)がんとRASopathy
ERK経路の上流に位置する分子(EGFR, Ras, Rafなど)は,膵・大腸・肺・甲状腺がん,悪性黒色腫などのさまざまな固形がんや,多発性骨髄腫,白血病を含む各種血液悪性腫瘍において高頻度に遺伝子変異が認められるがん遺伝子であり(表3),ERK経路を恒常的に活性化して発がんを導く65).特に,膵がんにおけるKRas変異(90%以上)や悪性黒色種におけるBRaf変異(50~60%)などのように,がん種特異的に高い変異率を示すがん遺伝子も同定されている66, 67).また近年の大規模がんゲノム解析から,MEK遺伝子の活性型変異が,低頻度ではあるものの肺・大腸・卵巣がん,悪性黒色腫など,さまざまながんで検出され68),MEKもがん遺伝子として機能することが明らかにされている.さらに,子宮頚がんや頭頚部がんではERK2の点変異も見いだされており69, 70),ホットスポット変異であるE322KはERK2のキナーゼ活性を亢進させることが証明されている71).
表3 ERKシグナル構成因子の各種がん組織における変異率 | 遺伝子 | 頻度 | 組織 |
|---|
| Ras | KRas | 90% | 膵がん |
| 50% | 大腸がん |
| 30% | 肺がん |
| 5% | 急性骨髄球性白血病 |
| NRas | 17% | 悪性黒色腫 |
| 14% | 急性骨髄球性白血病 |
| 6% | 甲状腺がん |
| HRas | 7% | 膀胱がん |
| 5% | 頭頚部扁平上皮がん |
| 2% | 甲状腺がん |
| MAPKKK | BRaf | 60% | 甲状腺がん |
| 50% | 悪性黒色腫 |
| 10% | 大腸がん |
| 6% | 肺がん |
| MAPKK | MEK | 7% | 悪性黒色腫 |
| 3% | 胆管がん |
| 2% | 大腸がん |
| 1% | 肺がん |
| MAPK | ERK | 8% | 子宮頚がん |
| 6% | 頭頚部扁平上皮がん |
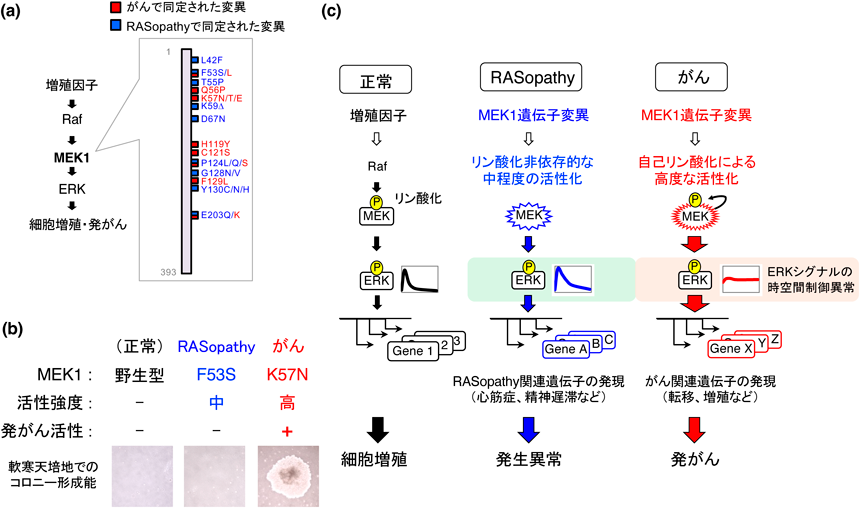
加えてERK経路構成因子の遺伝子変異は,がんのみならず常染色体優性遺伝性疾患である先天性Ras-MAPK症候群(RASopathy)の原因となることも明らかにされている72).RASopathyは,Costello症候群,Noonan症候群,Cardio-facio-cutaneous(CFC)症候群,LEOPARD症候群,神経線維腫症1型(NF1)など,多くの類似した臨床所見(頭蓋顔面異形,神経認知障害,肥大型心筋症,皮膚・筋骨格系異常など)を示す先天性疾患の総称であり,どの疾患においてもERK経路構成因子(SOS1, Ras, Raf, MEKなど)のいずれかに生殖細胞変異が検出される73).これらの知見は,ERK経路を中心とした生体内情報伝達が,組織や人体の正常な発生にもきわめて重要であることを示している.しかしながら,RASopathyの病態形成機構にはいまだ不明な点が多い.たとえば,MEK1遺伝子の点変異はCFC症候群患者の約25%に認められ,ERK活性を亢進させることが報告されているが,その一方で,CFC症候群患者に特に易発がん性は認められない.すなわち,がんおよびRASopathyにおいては,共通の遺伝子(RafやMEKなど)に点変異が見いだされるにもかかわらず,発がんおよび発生異常という異なる臨床像が形成されている.同一の遺伝子に変異がありながら,この表現型の違いがどのようにして生み出されているのか,その分子機構の詳細な解明はRASopathyの病態のみならず,発がん機構を理解する上でもきわめて重要であると思われる.
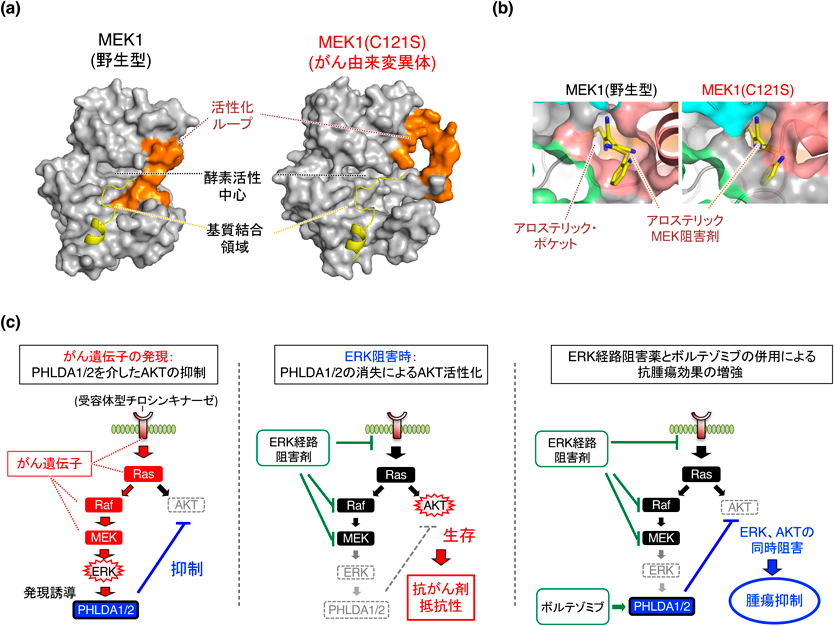
筆者らは最近,疾患由来MEK1点変異体の詳細な解析から,この問題の解明につながる知見を得ることに成功した74).我々はまず,がんとRASopathyの両疾患で,MEK1遺伝子の変異部位やアミノ酸変化の種類が異なることに着目し(図6a),これらのMEK1変異体に何らかの疾患特異的な性質の違いが認められるか検証を行った.MEK1は通常,増殖因子などの刺激に応じて上流のRafによってリン酸化されることで活性化するが,我々の解析から疾患由来MEK1変異体は,そのすべてが無刺激の状態でも高い酵素活性を示す恒常的活性化型となっていることが確認された.さらに興味深いことに我々は,各変異体の活性強度や性質が,原疾患に依存して大きく異なっていることを見いだした(図6b, c).すなわち,がん由来のMEK1変異体は,異常な自己リン酸化能(自分自身をリン酸化して活性化する能力)を獲得しており,きわめて強い酵素活性と細胞がん化能(悪性形質転換能)を有するのに対し,RASopathy由来の変異体は自己リン酸化能を有しておらず,リン酸化非依存的に活性化して中程度の酵素活性を示し,発がん能にも乏しいことを発見した.そこでさらにMEK1変異体の異常な活性化機構を解明するため結晶構造解析を実施した結果,RASopathy由来MEK1変異体では,活性化ループが分子の外側に向かって大きく開いており,これにより,野生型MEK1では同ループで隠されていた基質結合領域や酵素活性中心が解放されて,非リン酸化状態でも基質(ERK)をリン酸化しやすい状態になっていることがわかった(図7a).またこの変化に加えて,がん由来のMEK1変異体では,野生型MEK1で認められる近接した三つのアミノ酸残基(K57/H119/F129)間の相互作用(水素結合)が完全に消失しており,このことが異常な自己リン酸化能を獲得する主因であることを発見した.すなわち,この分子内水素結合の破壊によって,キナーゼ活性を抑制する作用を持つヘリックスA領域と酵素ドメイン間の相互作用が解除され,自己リン酸化能を獲得していることがわかった.実際に,これら三つのアミノ酸残基やその周辺領域の点変異は,がん患者において高頻度に認められることから,K57/H119/F129間の水素結合を破壊する変異を持つMEK1変異体のみが,自己リン酸化能を獲得して強力なキナーゼ活性を示し,最終的に発がんを導いていることが明らかとなった.
次に我々は,これら性質の異なる2種類のMEK1変異体が,細胞内でERKシグナルと遺伝子発現にどのような影響を与えるのか解析を行った.その結果,RASopathy由来の変異体を発現する細胞では,野生型細胞と比べて,無刺激時におけるERK活性の亢進は軽度であるものの,増殖因子刺激後に起こるERKの活性化と核移行が有意に増強し,その持続時間も延長することがわかった.またそれに伴って,細胞内のグローバルな遺伝子発現プロファイルが変化し,心筋症や精神遅滞などRASopathyの臨床症状に関連する遺伝子群の発現が選択的に亢進することを見いだした(図6c).一方,がん由来の変異体を発現する細胞では,無刺激の状態でもERKが強く活性化して核内に集積しており,その結果,特定の転写因子が常に活性化された状態となって発がんおよびがんの進展に関わる遺伝子が多数発現誘導されることを見いだした.これらの結果から,がんおよびRASopathyで認められるMEK1変異体の疾患特異的な性質の違いが,ERKシグナルの時空間制御と,細胞内の転写プログラムおよび遺伝子発現プロファイルに異なるタイプの異常を引き起こし,発がんと発生異常という別個の臨床像を導いていることが明らかとなった.
3)疾患由来MEK1変異体の解析から見いだされた新たな抗がん剤抵抗性獲得機構
各種がん遺伝子の機能を選択的に阻害し,ERK経路の異常な活性化を抑制する薬剤は,分子標的抗がん剤として活用されている.実際これまでに,EGFR阻害剤(erlotinib, gefitinib),KRas阻害剤(adagrasib, sotorasib),BRaf阻害剤(vemurafenib, dabrafenib),MEK阻害剤(trametinib, binimetinib)などが開発され,広く臨床の現場で使用されている2).これらの薬剤は一定の治療効果を示すものの,薬剤耐性の出現が大きな問題となっており,そのメカニズムの理解と克服は,より有効ながん治療の実現にきわめて重要である75, 76).上述の分子標的薬のうち,MEK阻害剤はMEK分子のみが持つアロステリック・ポケットと呼ばれる構造に結合する薬剤であることから,MEKに対する選択性がきわめて高く,現在,悪性黒色腫,非小細胞性肺がん,および結腸・直腸がんなどに対する治療薬として活用されているが,MEK遺伝子自体に変異を持つがんに対しては無効であることが知られている.その薬剤耐性機構の詳細はこれまで不明であったが,我々が実施したMEK変異体の構造解析から,点変異によってアロステリック・ポケットの形状が大きく変化して,阻害剤が結合不能となっていることが見いだされ,これが薬剤耐性獲得の原因であることが明らかとなった74)(図7b).その一方で,MEKの酵素活性中心であるATP結合ポケットの形状は,変異体においても変形せずに保たれていたことから,MEKに対する既存のATP競合阻害剤(hymenialdisine)を用いてその効果を検証したところ,変異体に対しても野生型と変わらず高い阻害活性を示すことが確認された.今後,変異型MEKを有するがんに対しては,ATP競合阻害剤の活用や,変形したアロステリック・ポケットの形状にも適合しうる新たな薬剤の開発が望まれる.
また筆者らは,上述の疾患由来MEK変異体を用いた遺伝子発現解析から,さまざまな分子標的抗がん剤(ERK経路阻害剤)に共通する,がんの新たな耐性獲得機構を見いだすことに成功した74).まず我々は,RASopathy由来のMEK1変異体(中程度のERK活性)では発現量が変化せず,がん由来のMEK1変異体(強力かつ恒常的なERK活性)を持つ細胞においてのみ,発現誘導される遺伝子が一定数存在することを見いだした.このような遺伝子はがん細胞特異的に発現しており,がんの病態に深く関与する可能性が想定される.興味深いことに,これらの遺伝子の中には,AKT阻害分子であるpleckstrin homology like domain family, member 1(PHLDA1)およびPHLDA2が含まれていた.そこでまず,実際のヒトがん組織におけるPHLDA1/2の発現を検証したところ,各種がん遺伝子(EGFR, Ras, Raf, MEK1等)に変異を有し,ERK活性の高いさまざまながん(肺・大腸・膵がん,悪性黒色腫など)で,PHLDA1/2がともに高発現していることが確認された.さらに,これらの分子の機能について解析を進めた結果,がん細胞におけるPHLDA1/2の高発現が,発がん過程とがん治療において「諸刃の剣」として機能していることがわかった.すなわち,PHLDA1/2は,元来,細胞の生存シグナル(AKT経路)を阻害する作用を持つ分子であり,ERK活性の高いがんで高発現することで,がん細胞の生存を抑制して腫瘍の増大を一定程度抑えているが(図7c),その一方で,がん治療の際に分子標的抗がん剤(ERK経路阻害剤)を患者に投与すると,がん細胞内のERK活性が低下してPHLDA1/2の発現が急速に消失してしまうため,これらの分子による抑制が解除されてAKT(生存)経路が強く活性化し,がん細胞が細胞死を回避して抗がん剤の効果が減弱してしまうことがわかった.実際に,がん細胞内にPHLDA1/2を強制的に導入してその発現を維持してやると,分子標的薬投与後もAKT活性が抑制されたままとなり,薬剤の抗腫瘍効果が著しく高まることが動物実験で確認された.この結果は,PHLDA1/2の発現を人工的に制御することができれば,分子標的抗がん剤の効果を飛躍的に高めることが可能であることを示している.そこでさらに,PHLDA1/2の発現を,ERK活性非依存的に亢進させる薬剤が存在するか探索を行った結果,既存薬ボルテゾミブが,転写および転写後調節の両レベルでPHLDA1/2の発現を増強する作用を持つことを見いだした.また実際に,分子標的抗がん剤(ERK経路阻害剤)とボルテゾミブをがん細胞に同時に投与すると,ERK活性が低下してもPHLDA1/2の発現が維持されて生存(AKT)シグナルが抑制されたままとなり,抗がん作用(がん細胞の細胞死)が有意に高まることが確認された(図7c).