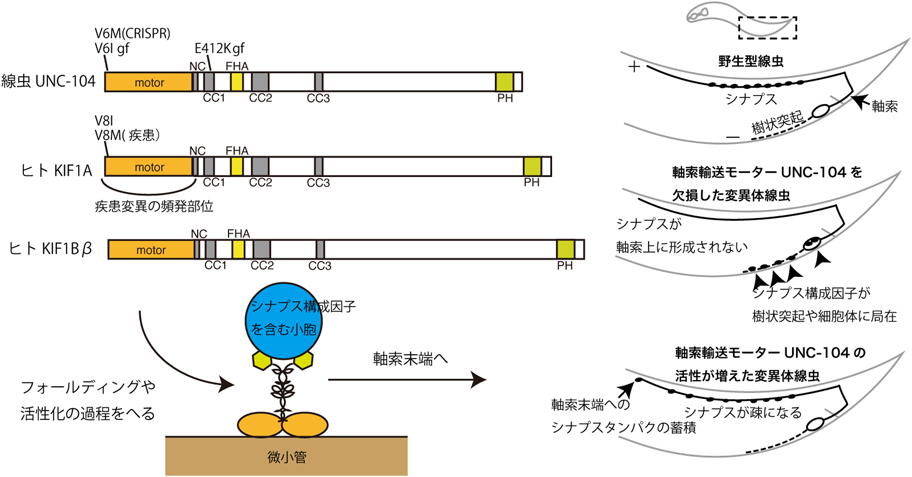
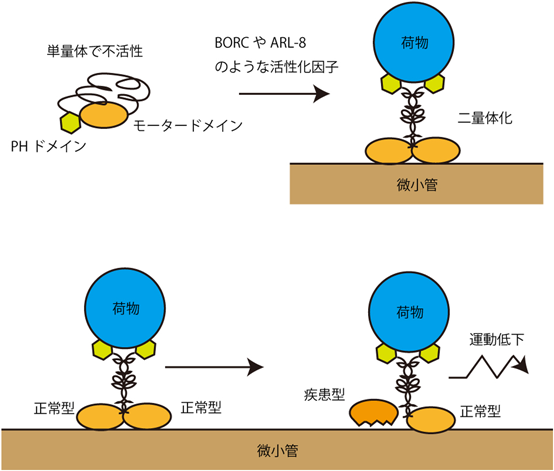
線虫遺伝学が明らかにする軸索輸送不全と神経変性Defects in axonal transport and neurological disorders revealed by worm genetics
東北大学学際科学フロンティア研究所Frontier Research Institute for Multidisciplinary Sciences, Tohoku University ◇ 〒980–0845 宮城県仙台市青葉区荒巻字青葉6–3 ◇ 6–3 Aramaki-Aoba, Aoba-ku, Sendai, Miyagi 980–0845, Japan
発行日:2023年10月25日Published: October 25, 2023