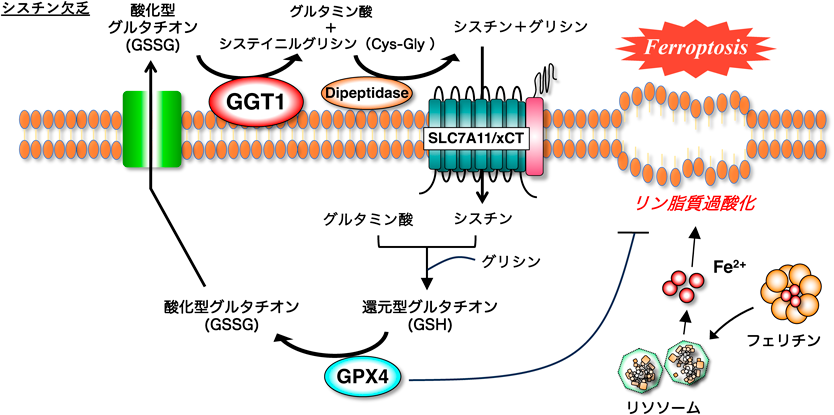
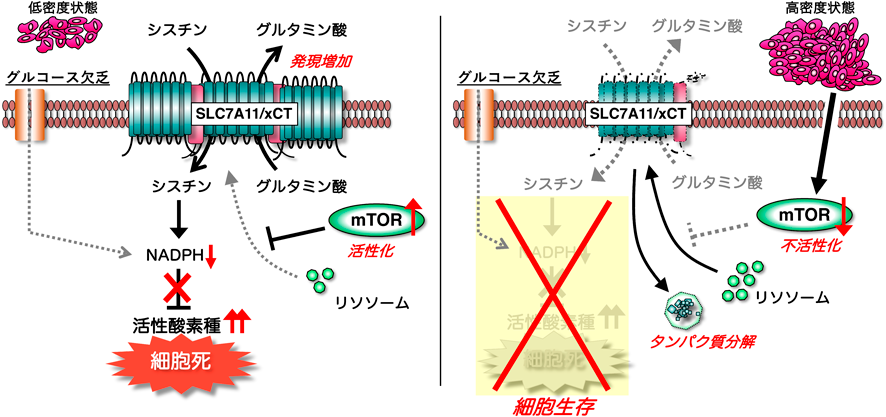
がん細胞のシスチン代謝に対する細胞外環境の影響Effects of the extracellular environment on cystine metabolism in cancer cells
1 京都大学大学院生命科学研究科生体システム学分野Laboratory of Molecular Neurobiology, Graduate School of Biostudies, Kyoto University ◇ 〒606–8501 京都市左京区吉田近衛町 ◇ Yoshidakonoe-cho, Sakyo-ku, Kyoto 606–8501, Japan
2 大阪公立大学大学院理学研究科生物化学専攻Department of Biological Chemistry, Graduate School of Science, Osaka Metropolitan University ◇ 〒599–8531 大阪府堺市中区学園町1–1 ◇ 1–1 Gakuen-cho, Naka-ku, Sakai, Osaka 599–8531, Japan
発行日:2023年12月25日Published: December 25, 2023