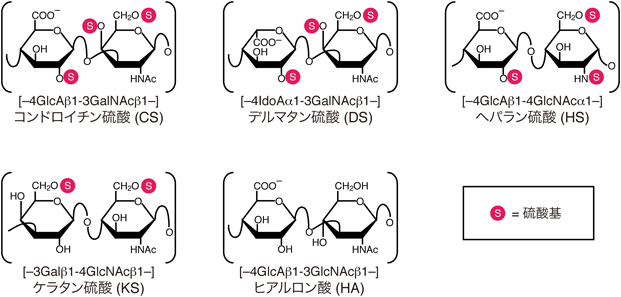
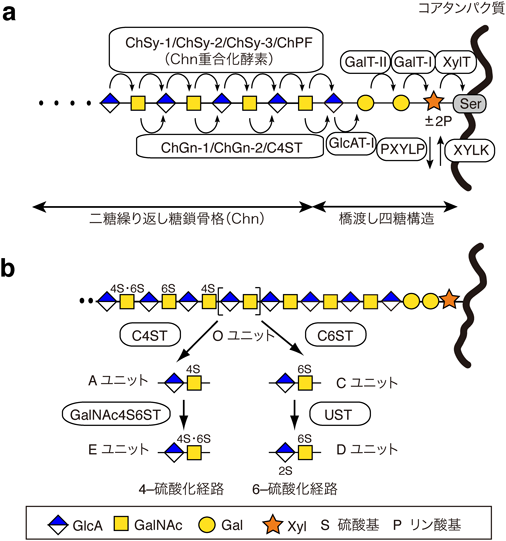
CS鎖の糖鎖骨格は,ウロン酸としてグルクロン酸(glucuronic acid:GlcA),アミノ糖としてN-アセチルガラクトサミン(N-acetylgalactosamine:GalNAc)を構成二糖単位とする繰り返し配列からなる(図1).この二糖繰り返し糖鎖骨格は,直接コアタンパク質に共有結合しているのではなく,コアタンパク質の特定のセリン(serine:Ser)残基上で合成された橋渡し四糖構造(GlcA-Gal-Gal-Xyl-Ser)を介して結合する[Xylはキシロース(xylose)を表す].この四糖構造は,小胞体・ゴルジ体に局在する4種の特異的な糖転移酵素によって,対応する単糖単位が順次転移されることで合成される(図2a)1–5).組み上がった四糖構造は,CS生合成のプライマーとなり,非還元末端のGlcA残基に,GalNAcとGlcAが交互に付加されることで,CS鎖特有の二糖繰り返し骨格(chondroitin:Chn)が合成される(図2a)6–8).面白いことに,HS鎖の二糖繰り返し糖鎖骨格の生合成もこの橋渡し四糖構造を起点に進行する.したがって,機能的なCS鎖やHS鎖を産生するためには,まずこの四糖構造が正しく生合成される必要がある.
CS鎖の二糖繰り返し糖鎖骨格の生合成には,コンドロイチン合成酵素(chondroitin synthase:ChSy)に代表される6種類の糖転移酵素,ChSy-19),ChSy-210, 11),ChSy-312, 13),コンドロイチン重合化因子(chondroitin polymerizing factor:ChPF11, 13, 14)),コンドロイチンGalNAc転移酵素(chondroitin GalNAc transferase:ChGn-116–18),ChGn-218, 19))が参画する(図2a).これら酵素のうち,前者四つの酵素(ChSy-1, ChSy-2, ChSy-3, ChPF)は,いずれか二つの酵素を組み合わせることで,in vitroにおいても二糖繰り返し糖鎖骨格(Chn)の重合化を促進することから12–14),“Chn重合化酵素”としての機能を果たすと考えられる(図2a).一方,後者二つの酵素(ChGn-1, ChGn-2)は,Chnの重合化活性を示さず,むしろCS鎖の鎖長やコアタンパク質に結合するCS鎖の本数を制御することによって,CS鎖の発現量を微調整する役割を担う20, 21).このようなChGnを介するCS鎖の鎖長や本数の制御には,後述する特定の硫酸化酵素(C4ST)との協調作用が不可欠である(図2a).
特筆すべきことに,CS鎖の合成量は,橋渡し四糖構造中のXyl残基の一過的なリン酸化によっても制御される(図2a).なぜなら,Xyl残基のリン酸化は,その後の連続したGalの転移とGlcAの転移を担う糖転移酵素活性を賦活化するためである22–24).筆者らの研究グループは,このリン酸化と脱リン酸化を触媒する責任酵素として,それぞれXyl 2-O-キナーゼ(xylose 2-O-kinase:XYLK)と2-O-ホスホキシロースホスファターゼ(2-O-phosphoxylose phosphatase:PXYLP)を同定し,Xyl残基の一過的なリン酸化による橋渡し四糖構造の成熟とCS鎖の合成量制御が,生理的に重要なイベントであることを証明した25–27).
3. CS鎖の硫酸化経路とその責任酵素の遺伝子欠損マウスの表現型
CS鎖の二糖繰り返し糖鎖骨格は,生合成過程で硫酸化やウロン酸の異性化などのさらなる修飾を受けて機能的な成熟体となる6–8, 28, 29).硫酸化修飾は,主にGlcA残基の2位やGalNAc残基の4位または6位のヒドロキシ基で起こる.したがって,硫酸化修飾後のCS鎖は,硫酸化パターンの異なる5種類の二糖単位(O, A, C, D, Eユニット)(図2b)の組合わせから構成されるポリマーと捉えることができる.また,異性化反応により,一部のGlcA残基がイズロン酸(iduronic acid:IdoA)残基へ構造変換された糖鎖骨格は,CS鎖の構造異性体であるDS鎖の前駆体に相当する(図1).DS型の糖鎖骨格もCS鎖と類似した硫酸化修飾過程を経て成熟体となる.DS鎖の生合成過程や機能の詳細については,紙面の制約上割愛させていただく.
CS鎖の硫酸化は,複数の硫酸基転移酵素の競合的あるいは協調的作用によって厳密に制御され,参画する酵素の基質特異性に基づき,「4-硫酸化経路」と「6-硫酸化経路」に大別される6–8, 28).図2bに示したように,両経路は,基本糖鎖骨格中のOユニット[GlcA-GalNAc](硫酸化されていない二糖ユニット)を共通の出発基質として,ある程度競合的に進行すると考えられている.まず,コンドロイチン4-O-硫酸基転移酵素(chondroitin 4-O-sulfotransferase:C4ST)30–33)によりOユニット中のGalNAc残基の4位が硫酸化されるとAユニット[GlcA-GalNAc(4-O-硫酸)]が,一方で,コンドロイチン6-O-硫酸基転移酵素(chondroitin 6-O-sulfotransferase:C6ST)34–36)によりGalNAc残基の6位が硫酸化されるとCユニット[GlcA-GalNAc(6-O-硫酸)]が合成される.さらに,Aユニットの一部がGalNAc 4-硫酸6-O-硫酸基転移酵素(GalNAc 4-sulfate 6-O-sulfotransferase:GalNAc4S6ST)37)によって硫酸化されるとEユニット[GlcA-GalNAc(4,6-O-ジ硫酸)]が,Cユニットの一部がウロン酸2-O-硫酸基転移酵素(uronyl 2-O-sulfotransferase:UST)38)によって硫酸化されるとDユニット[GlcA(2-O-硫酸)-GalNAc(6-O-硫酸)]が形成される.こうした生合成経路によって生み出されるCS鎖の構造多様性は,CS鎖が多彩な機能を発揮する原動力となっている.
実際,CS鎖における特異的な硫酸化構造が生理的に重要であることは,上述した硫酸基転移酵素群のシングルノックアウト(knockout:KO)マウスの表現型の違いからも明白である.「4-硫酸化経路」の最初のステップを担うC4STには,3種のファミリー分子(C4ST-1, C4ST-2, C4ST-3)が存在する7, 8).ジーントラップ法により得られたC4ST-1のKOマウスは,重篤な軟骨形成異常を呈し,呼吸困難により生後まもなく死に至る39).このマウスの組織では,4-硫酸化CS鎖の発現が野生型マウスの1割程度にまで減少する39)ことから,C4ST-1が「4-硫酸化経路」の主たる酵素であると考えられ,4-硫酸化CS鎖が少なくとも胎生期の骨格形成過程に重要であることを示唆する.その他の硫酸化ステップは,それぞれ1種類の酵素によって担われている.「6-硫酸化経路」の最初のステップを実質的に触媒するC6ST-1のKOマウスは,C4ST-1 KOとは対照的に,正常に生まれ,脾臓におけるTリンパ球の顕著な減少を除いて,見かけ上大きな異常はないとされた35).しかしながら,最近の研究成果から,C6ST-1のKOは,皮膚の肥厚とバリア機能の低下を招き,乾癬発症の危険因子になることが見いだされた.この発見により,皮膚の恒常性維持における6-硫酸化CS鎖の重要性が注目されている40).GalNAc4S-6ST KOマウスの組織では,Eユニットを完全に欠失したCS鎖が得られる41).一方で,当該マウスの表現型解析では,報告当初,マスト細胞におけるプロテアーゼ活性の減弱を除き,明確な発達異常は観察されていなかった41).しかしながら,その後の詳細な解析により,骨密度の顕著な低下が認められている42).その主な原因が骨芽細胞の分化不全であることも判明している42).
4. 神経可塑性の制御におけるCS鎖の硫酸化バランスの重要性
上述したように,CS鎖の硫酸化は,「4-硫酸化経路」と「6-硫酸化経路」のバランスによって成り立っている.したがって,CS鎖の4-硫酸化と6-硫酸化の割合を示す“4S/6S比”は,CS鎖の硫酸化状態を評価する指標としてしばしば用いられてきた.そこで本節では,CS鎖の硫酸化修飾の重要性を裏づける画期的な発見へとつながった,CS鎖の4S/6S比と神経可塑性との関連について紹介する.
神経可塑性とは,さまざまな外的または内的刺激に適応して,神経回路を再編成する性質のことをさす.幼少期の神経回路網は,外部環境からの刺激(経験)に対して鋭敏に応答する.こうした経験依存的な神経可塑性は,“臨界期”と呼ばれる特定の時期で最も強くみられ,臨界期を過ぎると顕著に低下する.特筆すべきことに,臨界期の終了は,ペリニューロナルネット(perineuronal net:PNN)の形成と連動していることが知られている43–45).PNNは,カルシウム結合タンパク質であるパルブアルブミン(parvalbumin:PV)を発現する抑制性介在ニューロン(PV陽性ニューロン)の細胞体や近位樹状突起を取り囲むように形成される特殊な細胞外マトリックス構造体である43–45).コンドロイチン硫酸プロテオグリカン(chondroitin sulfate proteoglycan)はPNNに集積する主要な構成成分であり,これまでにもCSPGの神経可塑性への関与が報告されている.たとえば,視覚野における眼優位可塑性があげられる.臨界期の間に片眼のみを遮蔽すると,視覚野ニューロンの遮蔽した眼に対する応答性は失われ,反対に開いている眼に対する応答性が強化される.その結果,遮蔽した片眼の視力は恒久的に失われるが,成体期に片眼遮蔽をしても視力に影響しない.この現象は,視覚野の経験依存的な神経回路網の再編機構を理解するモデルとして広く利用されている46).興味深いことに,臨界期を過ぎた成体に,細菌由来のCS分解酵素であるコンドロイチナーゼABC(chondroitinase ABC:ChABC)を投与して,視覚野に発現するCSPGのCS鎖を分解除去すると,PV陽性ニューロン周囲のPNNが崩壊し,眼優位可塑性が再賦活化する47, 48).また,恐怖記憶の定着に関与する扁桃体のPNNをChABCで狙い撃ちすることで,その後の恐怖記憶が消去できることも報告された49).これらの研究から,PNNの形成・維持や神経可塑性制御において,CSPGの糖鎖部分であるCS鎖の重要性が認知されるようになったが,その硫酸化構造の意義については見過ごされてきたきらいがある.なぜなら,ChABCを利用してCS鎖の機能を探る研究では,あらゆる硫酸化構造のCS鎖が分解除去されてしまっているためである.
CS鎖の4S/6S比は,脳の発達過程で劇的に変動することが報告されている50–52).一般に,CS鎖中の6-硫酸化の割合が発達に伴い次第に低下するのに対し,4-硫酸化の割合は増加する傾向を示す.つまり4S/6S比は,発達に伴い実質的に増加する.興味深いことに,筆者らの研究グループが作出したヒトのC6ST-1を過剰発現するトランスジェニック(transgenic:Tg)マウスは,「6-硫酸化経路」が亢進するため,相対的に4S/6S比が低く,成体期においても幼若期と同等レベルの眼優位可塑性を保持していることが判明した53).このマウスでは,PNNの形成低下に加え,本来PV陽性ニューロンの周囲にみられるOrthodenticle homeobox 2(Otx2)の集積低下も観察された53).Otx2は,PV陽性ニューロンの成熟過程に貢献し,眼優位可塑性を制御するホメオタンパク質として機能する54).したがって,脳の発達に伴うCS鎖の4S/6S比の増加は,Otx2を介したPV陽性ニューロンの成熟を調節することで,神経可塑性を担保する臨界期の終了時期を制御している可能性が考えられる53, 55).この事象においてCS鎖は,Otx2の補受容体もしくはリザーバーとしての役割を果たしているかもしれない.また,PNNに局在する代表的なCSPGの中にアグリカンがある.PNN中のアグリカンはADAMTS(a disintegrin and metalloprotease with thrombospondin motifs)などのタンパク質分解酵素によって代謝され,その安定性は,4S/6S比が相対的に低い若齢型のCS鎖を持つものほど低下することもわかった56).したがって,CS鎖の硫酸化バランスは,Otx2の機能制御のみならず,アグリカンをはじめとするCSPGの安定性に影響を与えることで,複合的にPNNの形成や神経可塑性を制御していると考えられる.
こうしたCS鎖の4S/6S比の上昇はPNNの加齢性変化に関わり,神経可塑性を低下させるため,CS鎖の硫酸化をターゲットとして加齢に伴う記憶力の低下や認知機能の低下の改善にも応用できるかもしれない.実際,野生型マウスに比べ,4S/6S比が高いC6ST-1 KOマウスでは,若齢段階から記憶障害が顕在化する傾向にある57).さらに重要なことは,老齢個体で6-硫酸化レベルを回復させると,加齢に伴う記憶障害や記憶機能に深く関わる嗅周皮質における長期増強が回復したことである57).したがって,特にCS鎖の「6-硫酸化経路」の賦活化は,加齢に伴う記憶障害の改善を目指す上で有望な戦略の一つであろう.
5. 遺伝性骨硬化症の発症につながるCS鎖の硫酸化バランス制御
繰り返しになるが,CS鎖の4S/6S比は,「4-硫酸化経路」と「6-硫酸化経路」のバランスによって構築される.したがって,各経路の律速段階に相当するC4STとC6STの発現レベルを調節することが,細胞,組織,ひいては個体レベルでのCS鎖の4S/6S比を決定する上で有効な手段であることは,神経可塑性制御の節でふれたとおりである.
特筆すべきことに,筆者らの研究グループの最近の研究成果から,この二つの硫酸化経路の選択が,C4STとC6STの発現レベルといった量的な調節のみならず,当該硫酸化酵素の活性制御による質的な調節によっても制御されていることが判明した58).その質的な調節を担う分子は,なんと四糖橋渡し構造中のキシロース残基のリン酸化を担うXyl 2-O-キナーゼ(XYLK;FAM20BおよびFAM20C)であった.
本稿の締めくくりとしてその調節機構の発見に至った経緯と,遺伝性骨硬化症の発症との関連について概説する.
1)Raine症候群の原因遺伝子:FAM20C
骨組織の発達とそれに続く恒常性の維持は,骨芽細胞による骨形成と破骨細胞による骨吸収のバランスによって担保されている.このバランスの破綻は,骨粗鬆症や骨硬化症などのさまざまな骨系統疾患の発症につながる.Raine(レイン)症候群は,FAM20C遺伝子の変異を原因とする常染色体潜性の遺伝性疾患で,頭蓋顔面奇形をはじめ,骨格の変形,組織の形成異常などを主徴とする致死性の骨硬化症である59–61).患者の表現型から,FAM20Cは,骨形成やバイオミネラリゼーションを負に制御するタンパク質であることが想定された.ところがFam20cのKOマウスは,むしろ逆の骨軟化様症状を呈する62, 63).その後,非致死性のRaine症候群として報告された患者では,低リン血症性くる病や骨軟化症を示すケースも見受けられている61).しかしながら,致死性の患者とFam20c KOマウスの表現型に大きな違いが生じる原因については依然として未解決のままであった.
2)カゼインキナーゼとしてのFAM20C
ミルクに含まれるカゼイン(casein)が高度にリン酸化されていることは古くから知られている.カゼインは,in vitroにおいても実に多くのプロテインキナーゼによってリン酸化される.しかしながら,いずれも細胞質に存在するキナーゼによる非特異的な修飾であり,カゼインなどの分泌タンパク質のリン酸化を触媒する真の酵素の実体は長い間謎であった.FAM20Cは,まさにこうした条件に合致する,小胞体やゴルジ体などの分泌経路に局在する生理的なカゼインキナーゼ(casein kinase)として世界的に注目を集めた64, 65).網羅的な解析から,FAM20Cは,ミルクタンパク質や骨組織の細胞外マトリックスタンパク質のみならず,さまざまな組織に含まれるタンパク質のリン酸化を触媒することがわかった64–66).FAM20Cの基質タンパク質として同定された分子のうち,低分子インテグリン結合リガンド・N結合型糖タンパク質(small integrin-binding ligand N-linked glycoproteins:SIBLINGs)やリン調節ホルモンであるFGF23(fibroblast growth factor 23)は,リン酸化の有無によりバイオミネラリゼーションを正に制御したり,負に制御したりする性質を持つ.したがって,Raine症候群患者やFam20c KOマウスにおける表現型の差異は,こうしたバイオミネラリゼーションに関与するタンパク質群のリン酸化状態の不均衡によって生み出されていると考えられていた.
3)XYLKとしてのFAM20C
分泌経路におけるリン酸化は,タンパク質だけでなく糖残基においても起こる.2節で述べたように,GAG鎖の橋渡し四糖構造の生合成過程で,Xyl残基はリン酸化される(図2a).筆者らの研究グループは,FAM20Cのファミリー分子であるFAM20Bが,橋渡し四糖構造中のXyl残基のリン酸化を触媒するXYLK(Xylキナーゼ)であることをいち早く同定・報告していた25).3種のFAM20ファミリー分子(FAM20A, FAM20B, FAM20C)のうち,FAM20Bは進化系統樹上,他の二つの遺伝子の祖先にあたり,FAM20Cは,遺伝子重複などによりFAM20Bから生じた遺伝子である可能性が示唆されている67).こうした経緯に基づき,筆者らは,FAM20CもFAM20Bと同様に,橋渡し四糖構造のXyl残基のリン酸化に関与するXYLKとして働き,GAG鎖の生合成に貢献するという初期仮説を立てるに至った.そして,このような独自の観点から,FAM20Cの生理的役割と致死性のRaine症候群患者にみられる骨硬化症状との関連を探ることにした.
期待したとおり,FAM20Cは,カゼインキナーゼ活性のみならず,XYLK活性をも保持していた58).上述したように,Xyl残基のリン酸化は橋渡し四糖構造の合成を促し,GAG鎖の生合成を賦活化する.実際,FAM20Cを過剰発現させたHeLa細胞では,CS鎖およびHS鎖の産生量が有意に増加し,ノックダウンにより減少した.そこで筆者らは,FAM20CにおけるXYLK活性の消失が骨硬化症発症につながることを期待し,致死性のRaine症候群患者にみられる4種の変異型FAM20C(G379R, G379E, L388R, R549W)の組換えタンパク質を作製し,それらの酵素活性を調べた58).期待は見事に裏切られ,いずれの変異型FAM20Cも野生型と同等のXYLK活性を保持していた.また,野生型FAM20Cの過剰発現によりGAG鎖の産生は増加するが,各変異体を過剰発現させたHeLa細胞においてもGAG鎖の産生量は特段減少するわけでもなく,野生型FAM20Cの場合とほぼ同等の増加レベルであることがわかった.これらのことから,Raine症候群の発症要因は,FAM20CのXYLK活性に依存したGAG産生量の制御異常ではないと結論づけた.
4)FAM20CによるCS鎖の硫酸化バランス制御
変異型FAM20CがXYLK活性を保持していたことは,筆者らにとって意外な結果であったが,少なくともFAM20CがGAG鎖の産生量増加に寄与することについては確信を持つことができた.GAG鎖の産生量が増加する要因として,①橋渡し四糖構造の数が増えることによるGAG鎖の本数の増加と,②GAG鎖自体の鎖長の増大があげられる7, 8).両者の違いは,目的の検体から精製したGAG鎖をゲルろ過クロマトグラフィーにかけて,その鎖長分布を比較することで判別できる.上述したHeLa細胞におけるCS鎖を分析した結果,野生型FAM20Cの過剰発現によるCS産生量の増加要因には,①の本数の増加と,②の鎖長の増大,の両者が関係していると考えられた58).
古典的なCS鎖の生合成スキームから考えると,①の本数の増加は,FAM20CのXYLK活性依存的な効果であると捉えることができるが,②の鎖長の増大については,XYLK活性の関与を想定することが困難である.興味深いことに,変異型FAM20Cの過剰発現でCS産生量は増加していたが,②の鎖長の増大による効果が認めらなくなった.これらの知見から,FAM20CによるCS鎖長の増大はXYLK活性によるものではなく,FAM20Cの新たな機能的側面によるものと考えられた.
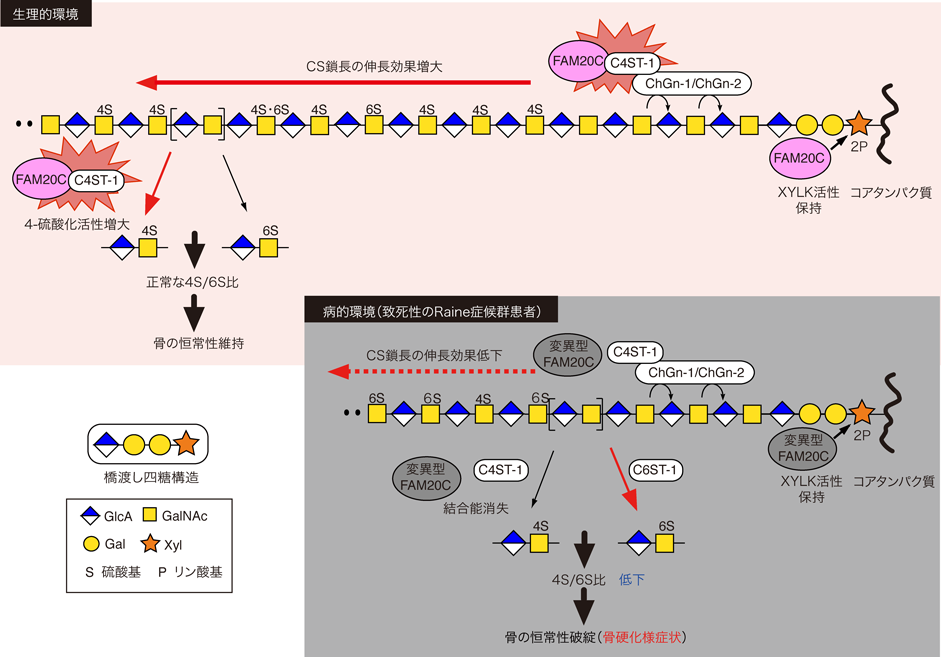
2節でも述べたように,CS鎖長の増大には,CS鎖の4-硫酸化が付随する7, 8).実際,C4ST-1を欠損したモデル細胞では,FAM20Cを過剰発現させてもCS鎖長の増大は観察されなかった58).このことから,FAM20CとC4ST-1における機能的連関について調べたところ,FAM20CがC4ST-1と相互作用し,C4ST-1の酵素活性を促進する働きを持ち合わせていることが新たに判明した58).その一方で,4種の変異型FAM20Cについては,C4ST-1との結合性やその4-硫酸化活性を高める効果のいずれも認められなかった58).これらの結果より,C4ST-1の活性制御能を失った変異型FAM20CがRaine症候群の発症と関連している可能性が考えられた.
C4ST-1の酵素活性がFAM20Cとの相互作用を介して直接的に制御されうることから,FAM20Cには,CS鎖の硫酸化バランス(4S/6S比)を調節する機能を兼ね備えていることが期待された.実際,FAM20Cを過剰発現させたHeLa細胞では,CS鎖の4S/6S比の顕著な増加が観察されたが,こうした変化は,変異型FAM20Cの過剰発現株では観察されなかった58).そこで筆者らは,バイオミネラリゼーションにおける変異型FAM20Cの影響を,CS鎖の硫酸化バランスの変化の観点から調べる計画を立てた.実験に供する細胞株の選定過程で,偶然にもヒト骨肉腫細胞株であるSaos-2細胞がFAM20C欠損株であることを見いだし,この細胞株を元にして,個々のFAM20タンパク質の安定発現株を樹立した.樹立した株におけるCS鎖の4S/6S比は,野生型FAM20Cの安定発現株で相対的に高値を示し,変異型FAM20Cの安定発現株においては有意に低値であった58).特筆すべきことに,この4S/6S比が低いほど,Saos-2細胞におけるバイオミネラリゼーションの程度が亢進する傾向を示した58).これらの結果より,致死性の変異型FAM20Cは,CS鎖の4S/6S比を低下させることによって,骨硬化を誘引している可能性が示唆された.
5)CS鎖における4S/6S比の低下によって誘引される骨硬化の分子基盤
CS鎖の4S/6S比の低下がなぜバイオミネラリゼーションを促進する原動力になるのであろうか? その分子基盤を探るために,マウス由来の骨芽細胞様株であるMC3T3-E1細胞を用いて実験的に4S/6S比を低下させることにした.その手段として,C4ST-1のノックダウン(またはKO)があげられよう.しかしながら,C4ST-1はCS鎖の産生量にも影響することから,この遺伝子の発現レベルを操作することは必ずしも最善策ではなかった.そのため,代替手段としてC6ST-1の過剰発現によるCS鎖の4S/6S比の改変を試みた.期待どおり,C6ST-1の過剰発現により,4S/6S比が顕著に低下した安定発現株が得られた58).また,これらの細胞では,親株やmock細胞に比べ,骨芽細胞分化やバイオミネラリゼーションの程度が有意に亢進することも判明した58).MC3T3-E1細胞の骨芽細胞分化は,N-カドヘリンおよびカドヘリン-11を介した細胞間接着により誘導される.これまでに筆者らの研究グループは,MC3T3-E1細胞が,Eユニットを多く含むCS鎖(「4-硫酸化経路」で生合成されるCS鎖で,CS-Eと呼ぶ)を多く産生する細胞であること,このCS-Eが,MC3T3-E1の細胞膜上に局在する両カドヘリン分子との相互作用を介して,骨芽細胞分化の促進に関与する下流シグナル伝達系を活性化することを証明している68).MC3T3-E1細胞のC6ST-1過剰発現株では,「6-硫酸化経路」が活発化するため,CS-Eの代わりにCユニットを多く含むCS鎖(CS-C)の産生が相対的に増加することになる.注目すべきことに,このCS-CもCS-Eと同様の挙動を示すこと,すなわち,両カドヘリン分子との結合を介して,下流の骨芽細胞分化誘導シグナル系を活性化してしまうことがわかった58).したがって,通常,発現レベルが低く保たれているCS-Cが,4S/6S比の低下により相対的に産生過多となる条件下では,変則的にカドヘリンシグナルが惹起され,非生理的なバイオミネラリゼーションが促進されてしまうモデルが想定された.
さらに興味深いことに,4S/6S比の低下を表現型に持つC6ST-1 Tgマウスの骨髄より採取した間葉系幹細胞の骨芽細胞分化能や長骨の骨密度を調べたところ,野生型と比較して,いずれも有意に高いことがわかった58).これらの知見をまとめると,4S/6S比の著しい低下は,致死性のRaine症候群患者において骨硬化様症状を顕在化させる主要因であることが示唆された.
6)FAM20Cの骨形成過程における役割と病態発現のカラクリ
本節の最後に,CS鎖の硫酸化バランスを調節するFAM20Cの機能的側面から,FAM20Cの骨形成過程における生理的役割と,Raine症候群の病態発現に至るカラクリについて考察したい(図3).本節でふれたように,FAM20Cは,C4ST-1との相互作用を通じてCS鎖の「4-硫酸化経路」をサポートする機能を果たし,CS鎖長の増大に基づくCS産生量の増加とともに4S/6S比の増加を担保する.これまでに筆者らの研究グループは,「4-硫酸化経路」で生合成されるCS-Eが,エストロゲンによって制御される骨形成過程において,必要不可欠な因子であることも見いだしている42).これらの事実を考え合わせると,生理的条件下におけるFAM20Cの機能は,骨組織におけるCS-Eの持続的な産生を支え,骨形成過程を正常に維持することにあると考えることができる(図3).対照的に,致死性の変異型FAM20Cに占拠された細胞環境では,C4ST-1に対するサポート機能がなくなるため,CS鎖における4S/6S比の低下を招く.その結果相対的に増加した6-硫酸化CS鎖(CS-C)は,非生理的なバイオミネラリゼーションを惹起して,骨硬化症状が顕在化すると考えられる(図3).特筆すべきことに,FAM20Bと変異型FAM20Cは,XYLK活性や,4S/6S比,バイオミネラリゼーションに与える影響など,筆者らが調べたCS生合成システムと骨形成パラメータに関するすべての項目において,類似した挙動を示した58).FAM20Bは乾癬の発症要因となるリスク遺伝子であることが報告されている69).また,3節でも述べたC6ST-1 KOマウスの皮膚における表現型から,6-硫酸化CS鎖の発現低下も乾癬の罹患率と相関すること40)を考慮すると,FAM20Bは何らかの形で「6-硫酸化経路」の活性化に貢献していると考えられる.これらのことから,変異型FAM20Cによって引き起こされる4S/6S比の低下は,変異型FAM20CがFAM20B様の機能を獲得した結果であるかもしれない.
Fam20cのKOマウスが骨軟化様症状を示す理由については,野生型FAM20Cによる生理的な骨形成促進能が破綻したためと考えればつじつまがあう.CS鎖の4S/6S比は,Fam20c KOマウスにおいても野生型に比べて低下するはずであるが,おそらくは,C6ST-1 TGマウスのように,「6-硫酸化経路」を積極的に活性化した環境が整わない限り,致死性のRaine症候群患者で観察されるような骨硬化症状は再現されないと予想している.あくまで筆者らの経験則ではあるが,マウスの細胞や組織由来のCS鎖は,概してヒト由来のCS鎖と比較して6-硫酸化の割合が低い傾向にある.したがって,変異型FAM20Cにより「4-硫酸化経路」が減弱したとしても,マウスでは骨硬化性の非生理的な分化スイッチを入れるほどの4S/6S比の低下が得られないものと推察している.