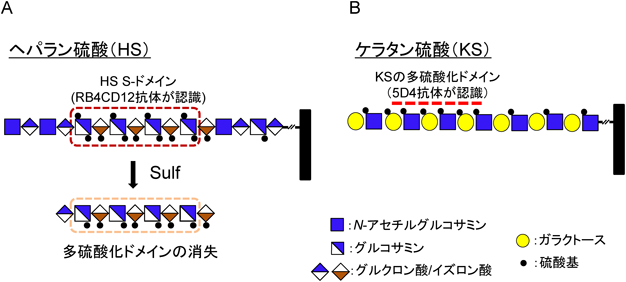
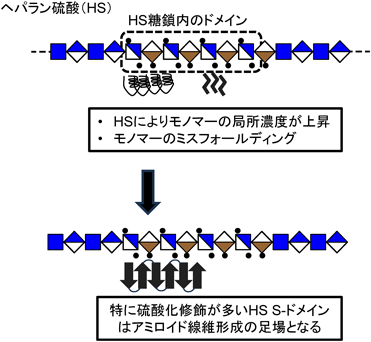
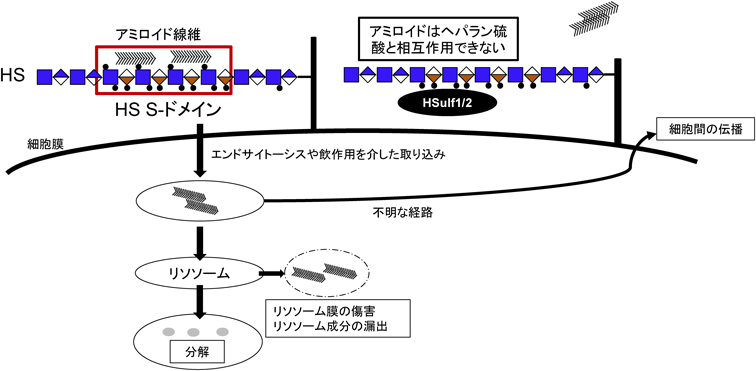
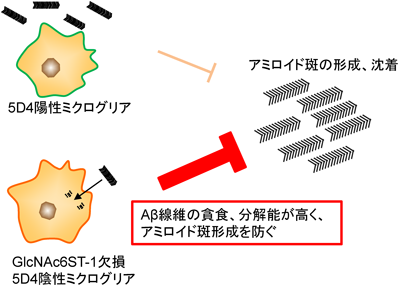
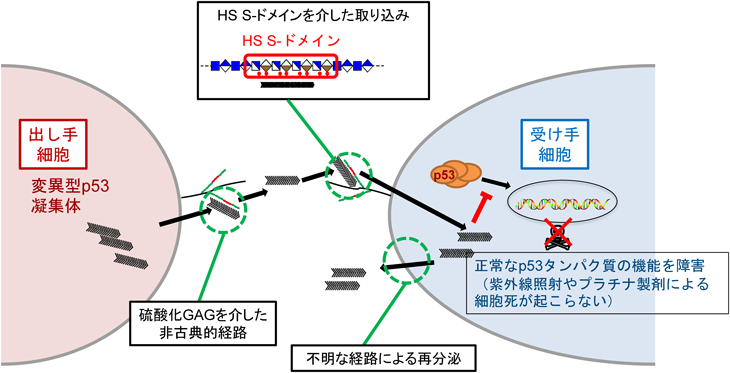
硫酸化修飾が制御するコンホメーション病におけるグリコサミノグリカンの病態機能Pathological roles of glycosaminoglycans in conformational diseases regulated by sulfation modifications
1 和歌山県立医科大学医学部生化学講座Department of Biochemistry, School of Medicine, Wakayama Medical University ◇ 〒641–8509 和歌山県和歌山市紀三井寺811–1 ◇ 811–1 Kimiidera, Wakayama 641–8509, Japan
2 フランス国立科学研究センター・リール大学糖鎖生物学構造機能研究所Unité de Glycobiologie Structurale et Fonctionnelle, CNRS, UMR 8576, Université de Lille ◇ 〒59655 ヴィルヌーブダスク メンドレーウヴ通りC9棟(フランス) ◇ Bat C9, Avenue Mendeleiev 59655 Villeneuve-d’Ascq, France
発行日:2024年4月25日Published: April 25, 2024