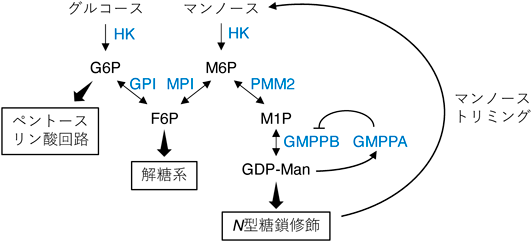
『生化学』の読者にとってマンノースの代謝経路はなじみが薄いかもしれない.そこで最初にマンノースの生合成と異化に関わる分子機構と生理的意義について概説したい.細胞に取り込まれたグルコースはフルクトース6-リン酸(fructose 6-phosphate:F6P)まで代謝されたのち,マンノースリン酸イソメラーゼ(mannose phosphate isomerase:MPI)の作用によってマンノース6-リン酸(mannose 6-phosphate:M6P)へと異性化される(図1).M6Pはさらにホスホマンノムターゼ2(phosphomannomutase 2:PMM2)およびGDP-マンノースピロホスホリラーゼB(GDP-mannose pyrophosphorylase B:GMPPB)によってGDP-マンノースへと代謝され,分泌経路においてタンパク質や脂質を修飾する糖鎖の生合成に利用される.マンノースの生合成に関わる一連の遺伝子(MPI, PMM2,およびGMPPA/B)は先天性糖鎖合成異常症(congenital disorders of glycosylation:CDG)の原因遺伝子である1).このことから,グルコースからマンノースを効率よく合成することは正常な糖鎖修飾に必要であり,生体にとってきわめて重要である.
糖鎖修飾を受けた分子が分泌経路を通過する間に,糖鎖はさまざまな糖加水分解酵素や糖転移酵素の働きによってその構造を大きく変化させる.この過程で生じる遊離のマンノース単糖は細胞外に分泌され,血中マンノースのプール(約50 μM)を形成する(図1)2).この血中プールはマンノースの摂取によって一過的に増加するが,全身の細胞によって取り込まれ,速やかに定常状態に戻る3).細胞に取り込まれたマンノースはM6Pに変換され,その一部はGDP-マンノースの生合成に再利用されるが,大部分はMPIの作用によってF6Pに変換され,解糖系で異化される.MPI活性をほとんど持たないミツバチにとってマンノースの摂取は致死的であり4),その原因は流入したマンノースがM6Pとして細胞内に異常に蓄積することによって引き起こされるグルコース代謝の阻害とATPプールの減少であると考えられている5, 6).このミツバチ症候群と呼ばれる代謝異常はMPI遺伝子を欠損した哺乳動物細胞にマンノースを大量に投与しても観察される現象であり,この場合は細胞増殖が抑制される5).これらのことから,外因性のマンノースを解糖系で効率よく異化することはエネルギー代謝と増殖という細胞の基本的な機能を保証するために必要である.
一見すべてが解明されたかのようにみえるマンノース代謝であるが,まだまだ面白い機能や分子メカニズムが潜んでいることが糖鎖生物学の枠を超えて明らかになってきた.次節ではまず筆者が研究対象とするタンパク質のアスパラギン結合型糖鎖修飾(N型糖鎖修飾)に焦点を当て,マンノースの生合成経路が関与するN型糖鎖修飾の型破りな制御機構を紹介し,がんの治療で問題となる抗がん剤抵抗性の改善への展開について議論する.最後に,我々が行った緻密な代謝解析からみえてきたミツバチ症候群の新たな側面について紹介し,がんの新しい治療法の開発の可能性を議論する.
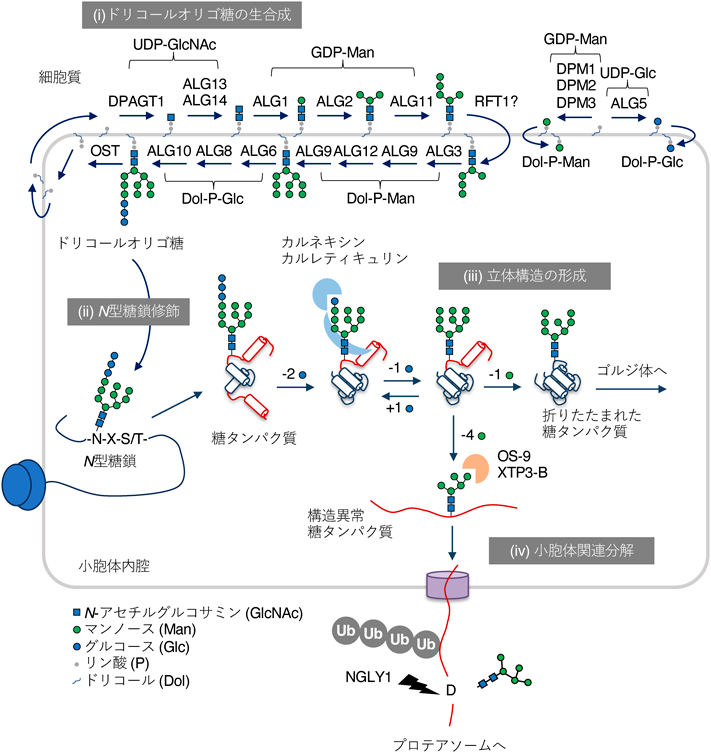
N型糖鎖修飾は小胞体で合成される分泌タンパク質や膜タンパク質中のコンセンサス配列(Asn-Xaa-Ser/Thr, XaaはPro以外のアミノ酸)に起こる基本的な翻訳後修飾の一つであり,これらのタンパク質の安定性や機能の調節を担う(図2)7).N型糖鎖修飾を触媒する酵素はオリゴ糖転移酵素(oligosaccharyltransferase:OST)であり,哺乳動物細胞では小胞体内腔に活性中心を持つSTT3AあるいはSTT3Bのいずれかを触媒サブユニットとする膜タンパク質複合体である.OSTの酵素反応はタンパク質の立体構造の形成と競合するにもかかわらず,N型糖鎖修飾の効率は非常に高い.このメカニズムについては本論から脱線するため,他の文献を参照されたい8).
N型糖鎖修飾の糖鎖供与体基質はドリコールオリゴ糖と呼ばれる糖脂質であり(図2),その構造は3個のグルコース(glucose:Glc),9個のマンノース(mannose:Man)および2個のN-アセチルグルコサミン(N-acetylglucosamine:GlcNAc)の計14糖からなる巨大な糖鎖(14糖鎖,Glc3Man9GlcNAc2)がピロリン酸を介してドリコール脂質(dolichol:Dol)に結合している(Glc3Man9GlcNAc2-PP-Dol).ドリコールオリゴ糖の生合成は小胞体膜の細胞質側で始まり,内腔側で終結する(図2,ステップi).まず,小胞体の細胞質側においてUDP-GlcNAcとドリコールリン酸からGlcNAc-PP-Dolが合成されると,さらに1分子のUDP-GlcNAcと5分子のGDP-マンノースが消費されてMan5GlcNAc2-PP-Dolが合成される.この中間体は小胞体膜の内腔側にフリップし,さらに4分子のDol-P-Manと3分子のDol-P-Glcが消費されてN型糖鎖修飾に至適な成熟型のGlc3Man9GlcNAc2-PP-Dolが合成される.最後にこの14糖鎖部分がOSTによってまるごとタンパク質に転移され,N型糖鎖が生合成される(図2,ステップii).小胞体においてタンパク質に付加された14糖鎖(Glc3Man9GlcNAc2)はさまざまな糖加水分解酵素によって順序よくトリミングされる過程で機能的に重要な糖鎖構造を露出させ,タンパク質の立体構造の形成を促進し(図2,ステップiii),一方で立体構造をうまくとれない異常なタンパク質を分解に導く,いわゆる糖タンパク質の品質管理機構の制御を担う(図2,ステップiv)7).
3. CDGにおけるドリコールオリゴ糖の分解と意義
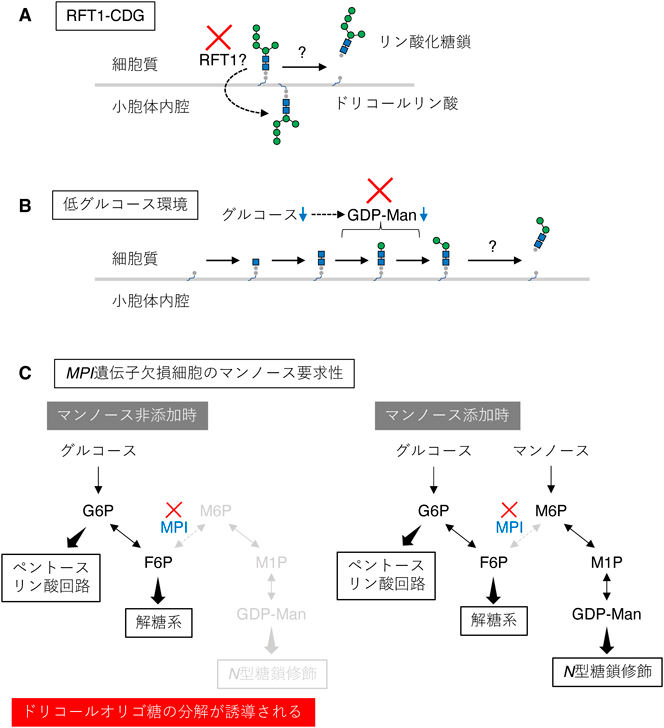
ドリコールオリゴ糖の生合成に関わるほぼすべての遺伝子がCDGの原因遺伝子として同定されている1).CDGはさまざまな糖鎖修飾の先天性異常の総称であるが,本稿ではドリコールオリゴ糖の生合成に異常を示すタイプのCDGを単にCDGと呼ぶ.通常,ドリコールオリゴ糖の生合成は一方向性に秩序立って進行するため,途中で異常な糖鎖構造が生じることなくGlc3Man9GlcNAc2-PP-Dolが合成される.しかし,CDGのように一つでも酵素反応が効率よく進まない状況ではその酵素の基質となるドリコールオリゴ糖中間体が蓄積し,成熟型のGlc3Man9GlcNAc2-PP-Dolが十分に合成されなくなる.OSTはGlc3Man9GlcNAc2-PP-Dolを最もよい糖鎖供与体基質とするため,CDGではN型糖鎖の付加効率が低下する.では,生成したドリコールオリゴ糖中間体はどうなってしまうのだろうか? 興味深いことに,CDG患者由来の線維芽細胞において生成したドリコールオリゴ糖中間体は未知の酵素によって速やかに分解され,リン酸化糖鎖とドリコールリン酸へと代謝されることが報告された(図3A)9, 10).CDGではこの分解経路を利用して新規合成に限りのあるドリコールを回収することで,ドリコールオリゴ糖の生合成が停滞するのを防いでいると考えられる11).しかし,細胞はCDGが起こることを見越してドリコールオリゴ糖の分解経路を用意しているとは考えにくい.
哺乳動物細胞や出芽酵母において,ドリコールオリゴ糖が分解されてリン酸化糖鎖が生じることは1970年代から知られていた12–14).しかし,リン酸化糖鎖の簡便な検出法がないことに加え,分解反応の生理的意義が不明であったために実験のアーティファクトとして長らく無視されていた.一方,我々は哺乳動物細胞を低グルコース培養するとドリコールオリゴ糖の分解が誘導され,短い糖鎖構造を持つリン酸化糖鎖が細胞質に蓄積することを見いだした(図3B)15).詳細なメカニズム解析の結果,グルコースの供給が乏しい環境ではGDP-マンノースが十分に合成されないため,ドリコールオリゴ糖の生合成がごく初期の段階で滞り,その結果,生成したドリコールオリゴ糖中間体がリン酸化糖鎖とドリコールリン酸へと分解されることがわかった15, 16).このとき,GDP-マンノースの合成不全がドリコールオリゴ糖の分解を誘導する直接的な原因であることを証明するために用いたMPI遺伝子欠損マウス由来胎仔線維芽細胞5)が,筆者とマンノース代謝との出会いであった.MPI遺伝子欠損細胞はグルコースからGDP-マンノースを合成できないが(図3C,左),培地に添加したマンノースからGDP-マンノースを合成できる(図3C,右).筆者はMPI遺伝子欠損細胞が示すマンノース要求性を逆手にとり,低マンノース培養によってGDP-マンノースの合成不全を誘導し,ドリコールオリゴ糖中間体がリン酸化糖鎖へと分解されることを証明した.
ドリコールオリゴ糖の分解酵素をコードする遺伝子が未解明であるため,低グルコース環境においてドリコールオリゴ糖が分解されることの意義も不明であるが,ここでは潜在的な機能を議論したい.一つはCDGでも提唱されているドリコールの再利用である.しかし,グルコース飢餓という一大事においてうまくいくはずのないドリコールオリゴ糖の生合成を何度も繰り返し行うためだけに蓄積した中間体を分解しているとは考えにくい.もう一つは糖鎖機能の保証である.低グルコース環境で生じるドリコールオリゴ糖中間体は糖タンパク質の品質管理機構に必要な糖鎖構造を欠いているため,不良品とみなされ,N型糖鎖修飾に利用される前に分解されているのかもしれない.このことから我々は,ドリコールオリゴ糖の分解経路は機能的な糖鎖だけがタンパク質に付加されるのを生合成の段階で保証する“糖鎖”の品質管理機構である可能性を提唱したが7, 15),この機構はドリコールオリゴ糖を分解してしまうため,N型糖鎖の付加効率が低下するという致命的な欠点がある.細胞の恒常性を維持するはずの品質管理機構が,恒常性を破綻しかねないリスクにもなるとはどういうことか? 現在,我々はこの矛盾点を解決し,型破りな制御機構の意義の解明を目指して研究を継続している.N型糖鎖の付加効率が低下すると立体構造を形成できない異常タンパク質が小胞体に蓄積し,小胞体ストレスが惹起され,unfolded protein response(UPR)が活性化することが容易に予想される.このことから,N型糖鎖修飾の効率を低下させるというリスクを冒してUPRを作動させるという,一見すると自暴自棄にも思える型破りな制御機構が存在する可能性がある.この点に関連して,MPI遺伝子の欠損が急性骨髄性白血病(acute myeloid leukemia:AML)の治療に有効であるという興味深い報告がなされた17).AMLにおいてFMS-like tyrosine kinase 3(FLT3)は患者の予後と関連する受容体型チロシンキナーゼであり,FLT3遺伝子に活性型internal tandem duplication(ITD)変異を有するAML患者は予後不良である.このFLT3-ITD変異に対する阻害剤がAMLの治療薬として開発されているが,薬剤耐性が問題となっている.この論文ではFLT3阻害剤に対するAML細胞の感受性に関連する遺伝子の網羅的探索において同定されたMPI遺伝子に着目し18),AML細胞のMPI遺伝子欠損によってFLT3阻害剤の効果が高まることが示された.まず,MPI遺伝子欠損により糖鎖修飾が抑制されると変性タンパク質が小胞体に蓄積する.これに応答してMPI欠損AML細胞はUPRのATF6経路を活性化し,遺伝子発現の変化を介して脂肪酸β酸化とATP産生を抑制する一方で,脂肪酸の取り込みと酸化ストレスを亢進させる.この一連の代謝変化は,MPI欠損が引き起こすN型糖鎖修飾の低下に対するAML細胞の生存戦略であることが示唆される.しかし,FLT3阻害剤は未知の機構を介してMPI欠損AML細胞への多価不飽和脂肪酸の取り込みをさらに増加させるため,脂質過酸化を介したフェロトーシスが誘導される.この脂質過酸化はATF6阻害剤の添加により抑制されることから,ATF6の活性化がFLT3阻害剤誘導性のフェロトーシスへのプライミングに重要である.特筆すべき点は,MPI遺伝子欠損が引き起こすFLT3阻害剤の効果の増強が血中濃度に相当するマンノースの添加によってもキャンセルされる点である.MPI欠損細胞にマンノースを添加するとGDP-マンノースが合成され,N型糖鎖修飾が回復するため,ATF6経路は作動しない.このことから,FLT3阻害剤に対するAML細胞の感受性の変化の起点はGDP-マンノースの欠乏によるドリコールオリゴ糖中間体の分解とそれに伴うN型糖鎖の付加効率の低下であると予想される.しかし,マンノースが恒常的に存在するマウスの血中においてもAML細胞のMPI遺伝子欠損によってFLT3阻害剤の効果が増強されることから,生体内ではGDP-マンノースの枯渇とは別にフェロトーシスへのプライミングを起こす機構の関与が示唆される.
M6PはGDP-マンノースの生合成に必須の中間代謝産物であるにもかかわらず,過剰に蓄積すると細胞毒性を示す.このため,細胞はMPIを使って余剰のM6PをF6Pに変換し,解糖系で異化(解毒)している.しかし,ミツバチのようにMPI活性が極端に低い個体やいくつかのがん培養細胞株ではマンノースの過度な流入によってM6Pが蓄積してしまう6, 19).MPI低発現(あるいは欠損)細胞におけるM6Pの蓄積メカニズムは未解明であるが,GDP-マンノースの合成量がGMPPAによって負にフィードバック調節されるため(図1)20),M6Pが効率よく下流に流れず蓄積すると考えられる.
ミツバチに対するマンノースの毒性にちなんで名づけられたミツバチ症候群は21),細胞内に過剰に蓄積したM6Pがグルコース代謝を阻害することによって引き起こされる代謝異常であり,哺乳動物細胞ではATPプールの減少と細胞増殖の抑制を特徴とする.M6Pが標的とするグルコース代謝酵素はヘキソキナーゼ,グルコースリン酸イソメラーゼおよびグルコース6-リン酸脱水素酵素であり5, 6),ミツバチ症候群ではエネルギー代謝とレドックス制御が破綻すると予想される.このようなミツバチ症候群に関わる分子メカニズムは主として試験管内における個別の酵素活性測定の結果に基づいて提唱されてきた経緯があり,細胞内で起こっている代謝異常の実態は不明であった.
この点を明らかにするために,我々はヒトがん細胞株においてMPI遺伝子を欠損させ,血中濃度の100倍に相当する過剰量のマンノース(5 mM)を投与することによってミツバチ症候群を誘導できるモデルを構築した22).このモデルを用いてエネルギー代謝解析を行った結果,過剰マンノースの添加とほぼ同時に解糖系が強力に抑制されるのに対し,ミトコンドリアにおける酸化的リン酸化(oxidative phosphorylation:OXPHOS)はわずかに活性化されるにとどまった.がん細胞は環境変化が著しい腫瘍内で生き延びるために好気的解糖系(ワールブルグ効果)をはじめとする代謝のリプログラミングを起こし,解糖系とOXPHOSのバランスを柔軟に変化させ,エネルギー代謝を最適化する環境適応機構を備えている23).しかし,ミツバチ症候群ではOXPHOSの活性化の度合いが低いため,解糖系の抑制に伴うATP産生量の損失を補うことができずにATPプールが減少することが判明した.本研究により,ミツバチ症候群におけるATPプールの減少のメカニズムの一端が明らかになったが,ミツバチ症候群のレドックス制御への影響は未解明であり,今後の課題である.
2018年,マンノースが抗腫瘍活性を持つという興味深い報告が英国のグループからなされた19).MPI低発現のがん細胞に対して25 mMという超高濃度のマンノースを作用させると,細胞増殖が抑制されるだけでなく,DNA障害型抗がん剤(シスプラチンやドキソルビシン;以下,単に抗がん剤と呼ぶ)の殺細胞効果が増強されるというのである.と,ここで一つの疑問が生じる.上述の抗がん剤はDNA複製阻害剤であり,増殖が盛んな細胞であるほど有効であると考えられる.では,マンノースはどのようにして増殖能が低下したがん細胞に対して抗がん剤の効果を増強できるのだろうか.
我々は,マンノースの抗腫瘍活性の発現に必要な条件がミツバチ症候群のそれと類似していることに着目し,先述のMPI遺伝子欠損ヒトがん細胞(以下,単にMPI欠損細胞)を用いてミツバチ症候群を誘導したところ,細胞増殖の抑制と抗がん剤の効果の増強が観察された22).ミツバチ症候群とマンノースの抗腫瘍活性との間に明確な関連性が示されたので,まずミツバチ症候群における細胞増殖の抑制機構の解明に取り組んだ.細胞増殖は細胞周期の進行によって制御される.そこで,細胞周期の蛍光プローブ[fluorescent ubiquitination-based cell cycle indicator(CA):Fucci(CA)]24)をMPI欠損細胞に導入してタイムラプス観察を行った結果,ミツバチ症候群ではFucci(CA)シグナルのパターンがきわめて複雑な多様性を示しており,そのすべてにおいて細胞周期の進行の著しい遅延が認められた.細胞周期と細胞内エネルギー代謝は双方向性に制御し合うことから25, 26),ミツバチ症候群におけるエネルギー代謝の変化が細胞周期の異常の一端を担っていると考えられる.さらに,ミツバチ症候群ではDNAの複製フォークは存在するもののDNAの複製が抑制されていたことから,複数のメカニズムによって細胞増殖が抑制されていることが示された.
次に,ミツバチ症候群による抗がん剤の効果の増強機構の解明に取り組んだ.細胞はDNA複製に先立ってG1期の間に数万から数十万箇所の複製起点に対して複製許可(ライセンス化)を行う.S期に移行するとライセンス化された複製起点のおよそ10%が活性化され,DNA複製が開始される27).残りの複製起点は“休眠”状態を保っているが,抗がん剤などの作用によって複製フォークの進行が止まると近傍の休眠複製起点が活性化し,DNA複製を継続させる.このバックアップ機構が破綻すると複製ストレスに対してゲノムが脆弱になることが知られている27–30).我々は,ミツバチ症候群が休眠複製起点からのDNA複製も抑制しているのではないかと予想し,抗がん剤の存在下において休眠複製起点を強制的に活性化した.その結果,ミツバチ症候群において休眠複製起点からのDNA複製が強力に抑制されることがわかった.これに伴い,ミツバチ症候群ではDNA損傷の中で最も致死性の高いDNA二重鎖切断のマーカーの発現が抗がん剤処理によって増加しており,ゲノムの脆弱化が認められた.以上のことから,ミツバチ症候群は平常時と複製ストレス時の両方においてDNA複製を抑制することが明らかとなった.
ミツバチ症候群の本質は細胞内に蓄積したM6Pが引き起こす代謝異常である.そこでミツバチ症候群によるDNA複製の抑制機構の手がかりを得るためにメタボロミクス解析を行ったところ,DNA合成の基質であるデオキシリボヌクレオシド三リン酸(deoxyribonucleoside triphosphate:dNTP)がミツバチ症候群で著明に減少していた.さらに,U-13C6-グルコーストレーサーを用いたグルコース炭素の代謝フラックス解析の結果,ミツバチ症候群ではプリン代謝およびピリミジン代謝へのグルコース炭素の流入が著しく低下しており,dNTPの新規合成が強力に抑制されていることがわかった.そこでdNTPの新規合成に関わるリボヌクレオチド還元酵素(ribonucleotide reductase:RNR)のRRM2サブユニットの阻害剤であるヒドロキシ尿素を用いてdNTPの新規合成経路を直接阻害してみると,ミツバチ症候群を誘導せずとも抗がん剤の効果が増強された.哺乳動物細胞にとってdNTP量を適切に保つことは正常なDNA複製だけでなく複製ストレスからの回復に必須であり,その破綻はゲノムの不安定化を引き起こす.以上のことから,dNTPの減少がミツバチ症候群の抗腫瘍作用の主要なメカニズムであることが示された.
dNTPのプールサイズは細胞周期およびDNA損傷依存的なdNTPの生合成と分解によって厳密に制御されており,その破綻はゲノムの不安定性を増大させる31, 32).ミツバチ症候群ではDNA損傷によって誘導されるRNRのRRM2Bサブユニット33)のタンパク質量が増加しており,タンパク質レベルではdNTP代謝の均衡が生合成に傾いている.それにもかかわらずdNTPが減少するのは,RNRの活性がATPによるアロステリック調節を受けるためにミツバチ症候群ではRNRの反応が起こりにくいからかもしれない34).また,ミツバチ症候群ではグルコース代謝が阻害されるため,グルコース炭素を必要とするプリン代謝およびピリミジン代謝の両方が抑制される.この代謝異常に関わる詳細なメカニズムは未解明であるが,興味深いことにプリン代謝ではリボース供与体基質(ホスホリボシルピロリン酸)の流入量が低下するのに対し,ピリミジン代謝では塩基を形成するアスパラギン酸の流入量が低下する.プリン・ピリミジン代謝はがんの治療標的として古くから注目されており,臨床試験中の薬剤を含めて多くの代謝拮抗薬や分子標的薬が開発されている.一方,ミツバチ症候群は多くのがん細胞で観察されるワールブルグ効果を標的とすることでプリン・ピリミジン代謝を抑制する点において既存の薬剤とは作用機序が一線を画す.今後,ミツバチ症候群によるプリン・ピリミジン代謝の抑制機構を解明することで,がんの新たな治療法の開発や治療標的の同定が可能になると期待される.
本稿では,我々の研究内容を中心に糖鎖生物学の枠を超えて広がりを見せ始めたマンノース代謝研究の最前線を紹介してきた.新たに同定されたマンノース代謝の機能はどれも細胞機能の根幹に関わるものばかりであり,基礎研究と応用研究の両面において今後の展開が期待される.一方で,未解決の問題も残されている.低グルコース環境においてGDP-マンノースの合成量が低下すると,なぜ哺乳動物細胞はリスクを冒してまでドリコールオリゴ糖を分解する必要があるのか? これは糖鎖の品質管理機構という概念だけでは到底説明がつかない.また,ミツバチ症候群に関する知見は代謝ネットワークの深層理解やその改変に大きな道筋を示すものであったが,そもそもM6Pはミツバチ症候群をどのように引き起こしているのだろうか? 今回紹介できなかったがM6Pはシアル酸の一つであるデアミノノイラミン酸(Kdn)35–37)や希少糖ヌクレオチドのUDP-マンノース38)にも代謝される.より詳細な分子メカニズムの解明とその応用に向け,ミツバチ症候群における代謝変化の全容を解明する必要がある.
謝辞Acknowledgments
筆者のラボメンバーとすべての共同研究者の方々に感謝申し上げます.
引用文献References
1) Lefeber, D.J., Freeze, H.H., Steet, R., & Kinoshita, T. (2022) Congenital Disorders of Glycosylation. in Essentials of Glycobiology (Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., Schnaar, R.L., & Seeberger, P.H., eds.), pp. 599–614, Cold Spring Harbor Laboratory Press, New York.
2) Sharma, V. & Freeze, H.H. (2011) Mannose efflux from the cells: A potential source of mannose in blood. J. Biol. Chem., 286, 10193–10200.
3) Alton, G., Kjaergaard, S., Etchison, J.R., Skovby, F., & Freeze, H.H. (1997) Oral ingestion of mannose elevates blood mannose levels: A first step toward a potential therapy for carbohydrate-deficient glycoprotein syndrome type I. Biochem. Mol. Med., 60, 127–133.
4) Staudenmayer, T. (1939) Die giftigkeit der mannose für bienen und andere insekten. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol., 26, 644–668.
5) DeRossi, C., Bode, L., Eklund, E.A., Zhang, F., Davis, J.A., Westphal, V., Wang, L., Borowsky, A.D., & Freeze, H.H. (2006) Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J. Biol. Chem., 281, 5916–5927.
6) Sols, A., Cadenas, E., & Alvarado, F. (1960) Enzymatic basis of mannose toxicity in honey bees. Science, 131, 297–298.
7) Harada, Y., Ohkawa, Y., Maeda, K., & Taniguchi, N. (2022) Glycan quality control in and out of the endoplasmic reticulum of mammalian cells. FEBS J., 289, 7147–7162.
8) Harada, Y., Ohkawa, Y., Kizuka, Y., & Taniguchi, N. (2019) Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. Int. J. Mol. Sci., 20, 6074.
9) Peric, D., Durrant-Arico, C., Delenda, C., Dupre, T., De Lonlay, P., de Baulny, H.O., Pelatan, C., Bader-Meunier, B., Danos, O., Chantret, I., et al. (2010) The compartmentalisation of phosphorylated free oligosaccharides in cells from a CDG Ig patient reveals a novel ER-to-cytosol translocation process. PLoS One, 5, e11675.
10) Vleugels, W., Duvet, S., Peanne, R., Mir, A.M., Cacan, R., Michalski, J.C., Matthijs, G., & Foulquier, F. (2011) Identification of phosphorylated oligosaccharides in cells of patients with a congenital disorders of glycosylation (CDG-I). Biochimie, 93, 823–833.
11) Rush, J.S., Cho, S.K., Jiang, S., Hofmann, S.L., & Waechter, C.J. (2002) Identification and characterization of a cDNA encoding a dolichyl pyrophosphate phosphatase located in the endoplasmic reticulum of mammalian cells. J. Biol. Chem., 277, 45226–45234.
12) Hsu, A.F., Baynes, J.W., & Heath, E.C. (1974) The role of a dolichol-oligosaccharide as an intermediate in glycoprotein biosynthesis. Proc. Natl. Acad. Sci. USA, 71, 2391–2395.
13) Cacan, R., Hoflack, B., & Verbert, A. (1980) Fate of oligosaccharide-lipid intermediates synthesized by resting rat-spleen lymphocytes. Eur. J. Biochem., 106, 473–479.
14) Belard, M., Cacan, R., & Verbert, A. (1988) Characterization of an oligosaccharide-pyrophosphodolichol pyrophosphatase activity in yeast. Biochem. J., 255, 235–242.
15) Harada, Y., Nakajima, K., Masahara-Negishi, Y., Freeze, H.H., Angata, T., Taniguchi, N., & Suzuki, T. (2013) Metabolically programmed quality control system for dolichol-linked oligosaccharides. Proc. Natl. Acad. Sci. USA, 110, 19366–19371.
16) Harada, Y., Huang, C., Yamaki, S., Dohmae, N., & Suzuki, T. (2016) Non-lysosomal degradation of singly phosphorylated oligosaccharides initiated by the action of a cytosolic endo-beta-N-acetylglucosaminidase. J. Biol. Chem., 291, 8048–8058.
17) Woodley, K., Dillingh, L.S., Giotopoulos, G., Madrigal, P., Rattigan, K.M., Philippe, C., Dembitz, V., Magee, A.M.S., Asby, R., van de Lagemaat, L.N., et al. (2023) Mannose metabolism inhibition sensitizes acute myeloid leukaemia cells to therapy by driving ferroptotic cell death. Nat. Commun., 14, 2132.
18) Gallipoli, P., Giotopoulos, G., Tzelepis, K., Costa, A.S.H., Vohra, S., Medina-Perez, P., Basheer, F., Marando, L., Di Lisio, L., Dias, J.M.L., et al. (2018) Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood, 131, 1639–1653.
19) Gonzalez, P.S., O’Prey, J., Cardaci, S., Barthet, V.J.A., Sakamaki, J.I., Beaumatin, F., Roseweir, A., Gay, D.M., Mackay, G., Malviya, G., et al. (2018) Mannose impairs tumour growth and enhances chemotherapy. Nature, 563, 719–723.
20) Zheng, L., Liu, Z., Wang, Y., Yang, F., Wang, J., Huang, W., Qin, J., Tian, M., Cai, X., Liu, X., et al. (2021) Cryo-EM structures of human GMPPA-GMPPB complex reveal how cells maintain GDP-mannose homeostasis. Nat. Struct. Mol. Biol., 28, 1–12.
21) Freinkel, N., Lewis, N.J., Akazawa, S., Roth, S.I., & Gorman, L. (1984) The honeybee syndrome—Implications of the teratogenicity of mannose in rat-embryo culture. N. Engl. J. Med., 310, 223–230.
22) Harada, Y., Mizote, Y., Suzuki, T., Hirayama, A., Ikeda, S., Nishida, M., Hiratsuka, T., Ueda, A., Imagawa, Y., Maeda, K., et al. (2023) Metabolic clogging of mannose triggers dNTP loss and genomic instability in human cancer cells. eLife, 12, e83870.
23) Sun, L., Suo, C., Li, S.T., Zhang, H., & Gao, P. (2018) Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg effect. Biochim. Biophys. Acta Rev. Cancer, 1870, 51–66.
24) Sakaue-Sawano, A., Yo, M., Komatsu, N., Hiratsuka, T., Kogure, T., Hoshida, T., Goshima, N., Matsuda, M., Miyoshi, H., & Miyawaki, A. (2017) Genetically encoded tools for optical dissection of the mammalian cell cycle. Mol. Cell, 68, 626–640.e5.
25) Icard, P., Fournel, L., Wu, Z., Alifano, M., & Lincet, H. (2019) Interconnection between metabolism and cell cycle in cancer. Trends Biochem. Sci., 44, 490–501.
26) Salazar-Roa, M. & Malumbres, M. (2017) Fueling the cell division cycle. Trends Cell Biol., 27, 69–81.
27) Blow, J.J., Ge, X.Q., & Jackson, D.A. (2011) How dormant origins promote complete genome replication. Trends Biochem. Sci., 36, 405–414.
28) Kawabata, T., Luebben, S.W., Yamaguchi, S., Ilves, I., Matise, I., Buske, T., Botchan, M.R., & Shima, N. (2011) Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell, 41, 543–553.
29) Shima, N., Alcaraz, A., Liachko, I., Buske, T.R., Andrews, C.A., Munroe, R.J., Hartford, S.A., Tye, B.K., & Schimenti, J.C. (2007) A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet., 39, 93–98.
30) Ge, X.Q., Jackson, D.A., & Blow, J.J. (2007) Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev., 21, 3331–3341.
31) Pajalunga, D., Franzolin, E., Stevanoni, M., Zribi, S., Passaro, N., Gurtner, A., Donsante, S., Loffredo, D., Losanno, L., Bianchi, V., et al. (2017) A defective dNTP pool hinders DNA replication in cell cycle-reactivated terminally differentiated muscle cells. Cell Death Differ., 24, 774–784.
32) Kumar, D., Viberg, J., Nilsson, A.K., & Chabes, A. (2010) Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res., 38, 3975–3983.
33) Tanaka, H., Arakawa, H., Yamaguchi, T., Shiraishi, K., Fukuda, S., Matsui, K., Takei, Y., & Nakamura, Y. (2000) A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature, 404, 42–49.
34) Brignole, E.J., Tsai, K.L., Chittuluru, J., Li, H., Aye, Y., Penczek, P.A., Stubbe, J., Drennan, C.L., & Asturias, F. (2018) 3.3-A resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. eLife, 7, e31502.
35) Kawanishi, K., Saha, S., Diaz, S., Vaill, M., Sasmal, A., Siddiqui, S.S., Choudhury, B., Sharma, K., Chen, X., Schoenhofen, I.C., et al. (2021) Evolutionary conservation of human ketodeoxynonulosonic acid production is independent of sialoglycan biosynthesis. J. Clin. Invest., 131, e137681.
36) Go, S., Sato, C., Furuhata, K., & Kitajima, K. (2006) Oral ingestion of mannose alters the expression level of deaminoneuraminic acid (KDN) in mouse organs. Glycoconj. J., 23, 411–421.
37) Angata, T., Nakata, D., Matsuda, T., & Kitajima, K. (1999) Elevated expression of free deaminoneuraminic acid in mammalian cells cultured in mannose-rich media. Biochem. Biophys. Res. Commun., 261, 326–331.
38) Nakajima, K., Kizuka, Y., Yamaguchi, Y., Hirabayashi, Y., Takahashi, K., Yuzawa, Y., & Taniguchi, N. (2018) Identification and characterization of UDP-mannose in human cell lines and mouse organs: Differential distribution across brain regions and organs. Biochem. Biophys. Res. Commun., 495, 401–407.
著者紹介Author Profile
 原田 陽一郎(はらだ よういちろう)
原田 陽一郎(はらだ よういちろう)大阪国際がんセンター研究所糖鎖オンコロジー部 チームリーダー.博士(農学).
略歴2001年静岡大学農学部卒業.03年同大学院農学研究科修了.07年名古屋大学大学院生命農学研究科修了.07~10年ストーニーブルック大学,10~15年理化学研究所,15~19年鹿児島大学大学院医歯学総合研究科を経て19年より現職.
研究テーマと抱負糖(鎖)の生合成と分解に関する基礎研究.糖に秘められた機能を明らかにしたい.
ウェブサイトhttps://researchmap.jp/harada_yoichiro
趣味海釣り.