生体では,各組織における実質細胞とさまざまな間質細胞との細胞間接着や液性因子を介した相互作用により誘導される可逆的な組織のリモデリングが,組織の恒常性維持機構として重要である1).各組織では,さまざまな内的・外的ストレスにより損傷が生じており,恒常性維持機構の適切な応答によりその損傷が修復され,恒常性が維持される1).ところが,長期的あるいは過度なストレスに曝露されることで,組織の恒常性維持機構の応答不全や過剰応答が引き起こされ,慢性炎症が惹起される1).通常,炎症反応は,感染に対する生体防御や損傷組織の修復の初期に一過性に生じ,病原体の排除や損傷治癒に伴い急速に収束する.一方,慢性炎症では,低レベルの炎症が長期間持続し,不可逆的な組織リモデリングが生じることで,組織や臓器の機能低下をもたらし,生活習慣関連疾患やがんなどさまざまな疾患の発症や,病態の進展にも寄与する1, 2).
アンジオポエチン様タンパク質(angiopoietin-like protein: ANGPTL)は,血管新生や幹細胞維持に関わるアンジオポエチンと類似の構造的特徴を有する分泌タンパク質である.ヒトでは,8種類(ANGPTL1~8)のファミリー分子が同定されており,マウスでは,ANGPTL5以外のファミリー分子が存在する3–5).ANGPTL8を除くANGPTLファミリー分子は,N末端側にコイルドコイルドメイン(coiled-coil domain: CCD),C末端側にフィブリノーゲン様ドメイン(fibrinogen-like domain: FLD)を有する3–5).ANGPTLは構造的にアンジオポエチンに類似しているものの,アンジオポエチンの受容体であるTie2やTie1には結合しない6).ANGPTLファミリー分子の多くは,integrinを受容体として作用するが,近年,integrin以外の受容体も報告されている4, 5).ANGPTLファミリー分子の生理的機能としては,その多くがアンジオポエチンと同様に,血管新生制御に関わっているが,これ以外にも糖・脂質代謝,エネルギー代謝,幹細胞制御,組織修復など,多彩な機能を示すことが報告されている3, 5, 7).我々はANGPTLファミリー分子のなかでも,特にANGPTL2に着目し,その生理および病態生理における機能解明を行ってきた.本稿では,ANGPTL2の生理的機能に加え,疾患発症・進展との関連について,特にがん病態における機能を中心に概説する.
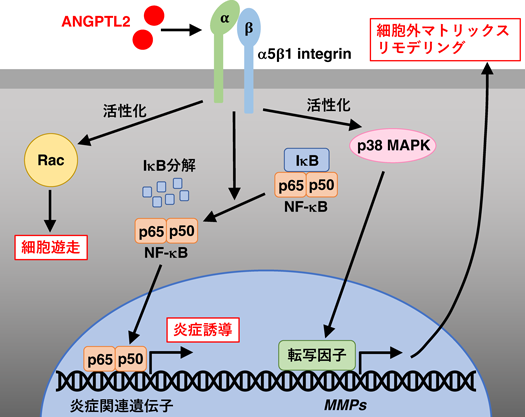
ゼブラフィッシュは,高い組織再生能を有するモデル生物である.我々は,ゼブラフィッシュの尾ヒレの損傷修復モデルにおいて,再生が活発になる時期に,脱分化した細胞が集合し,再生の起点となる芽体(blastema)で,Angptl2の発現が誘導されることを解明し8),ANGPTL2が組織修復に関与している可能性を見いだした.その後,デキストラン硫酸ナトリウム(dextran sulfate sodium:DSS)やブレオマイシンを用いた腸管や肺の組織損傷修復マウスモデルでの解析において,Angptl2ノックアウト(KO)マウスでは,野生型マウスに比べて組織損傷に対して修復不全が生じることが明らかとなった9, 10).このことから,ANGPTL2による組織の修復機構が種を超えて保存されており,生体の恒常性維持に重要な役割を担っていることが示唆された.ANGPTL2による組織修復機構として,ANGPTL2が血管新生,炎症経路活性化,および細胞外マトリックスのリモデリングなど,組織リモデリングを活性化することが明らかとなった4)(図1).ANGPTL2による組織リモデリングでは,その受容体であるα5β1 integrinを介したシグナル伝達経路が重要な役割を果たしており,血管新生では,α5β1 integrinを介して低分子量Rho GTPaseであるRacを活性化し,血管新生に重要な血管内皮細胞の遊走能を亢進する4, 11)(図1).また,α5β1 integrinを介してnuclear factor-κB(NF-κB)の抑制因子であるinhibitor of nuclear factor-κB(IκB)の分解を促進することで,NF-κBの核内移行を引き起こし,炎症関連遺伝子の発現を活性化すること4, 11),p38 mitogen-activated kinase(MAPK)の活性化を促進することで,matrix metalloproteinases(MMPs)の発現を活性化し,細胞外マトリックスのリモデリングに寄与することが明らかとなった4, 12)(図1).
組織リモデリング作用に加えて,腸管組織の損傷修復による恒常性維持機構では,ANGPTL2シグナルによる腸管上皮幹細胞の維持が重要であることを解明した5, 10).腸管組織の損傷修復では,腸管上皮幹細胞による上皮細胞の供給と幹細胞の自己複製のバランスが重要であり,これらは腸管上皮幹細胞において増殖・分化を促進するbone morphogenic protein(BMP)シグナルと幹細胞性維持に作用するWntシグナルのバランスで制御されている13, 14).BMPは,腸上皮下に存在する筋線維芽細胞によって産生されるが15),我々は,筋線維芽細胞からANGPTL2が産生されること,ANGPTL2がα5β1 integrinを介して筋線維芽細胞におけるBMPの産生量を調節していることを解明した5, 10).ANGPTL2によるBMPシグナルの抑制は,組織損傷部位における腸管上皮幹細胞の過剰な増殖・分化を抑制することにつながり,幹細胞性の維持に寄与することが明らかとなった5, 10).
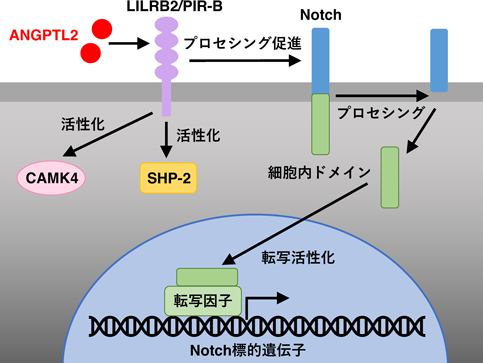
ANGPTL2シグナルは,アンジオポエチンと同様に,造血幹細胞の維持にも寄与していることが報告されている5, 16).種々の細胞において,α5β1 integrinがANGPTL2受容体として機能するが4),造血幹細胞では,leukocyte immune-like receptor B2(LILRB2)[マウスでは,paired immune-like receptor-B(PIR-B)]がANGPTL2の受容体として機能することが報告された17).ANGPTL2は,LILRB2/PIR-Bを介して,造血幹細胞の自己複製に関わるSH2 domain-containing protein tyrosine phosphatase-2(SHP-2)およびcalcium/calmodulin-dependent protein kinase 4(CAMK4)経路を活性化することや17)(図2),自己複製と幹細胞性維持に関わるNotchシグナルを活性化することなどが報告されている18)(図2).
ANGPTL2は,損傷修復による組織の恒常性維持に重要であるが,一方で,その過剰産生は,さまざまな疾患の発症や進展に寄与する4).生活習慣病では,ANGPTL2の過剰産生による慢性炎症が,病態発症・進展の基盤となっている4).たとえば,内臓脂肪組織においては,肥満病態が進行すると脂肪細胞でのANGPTL2産生が過剰となり,α5β1 integrin–NF-κB経路を介して血管内皮細胞の炎症経路活性化や血管の透過性亢進,さらにはマクロファージやT細胞の脂肪組織への浸潤が促進され,脂肪組織の慢性炎症および不可逆的な組織リモデリングを引き起こすことで,肥満から全身性インスリン抵抗性,糖尿病発症など,代謝異常を伴う肥満症への病態進展を促進する4, 11, 19).また,肥満をはじめとする心血管病発症の高リスク病態では,血管内皮細胞におけるANGPTL2シグナルを介した血管組織の慢性炎症,不可逆的な組織リモデリングが,血管内皮機能障害や動脈硬化性プラーク形成促進にも寄与する20, 21).さらに,慢性腎臓病の病態では,尿細管上皮細胞においてANGPTL2の過剰産生が生じ,その結果,α5β1 integrinを介してextracellular signal-regulated kinase(ERK)が活性化されることで,組織線維化の鍵因子であるtransforming growth factor-β(TGF-β)の発現が誘導される22).さらに,ANGPTL2により腎組織内へのマクロファージ浸潤が促進されることで炎症が遷延化し,結果として腎線維化を促進する22).
慢性炎症や組織リモデリングは,発がんやがんの浸潤・転移に関連することから,過剰なANGPTL2シグナルは,生活習慣病だけでなく,がん発症・進展にも共通する分子基盤として重要であると考えられた.我々は,がん病態におけるANGPTL2の機能を検討し,さまざまな種類のがんにおいて,ANGPTL2が病態促進に作用することを解明してきた.しかし,近年,ANGPTL2は,がん病態促進だけでなく,抑制にも作用する二面性があることが明らかとなってきた.以降の節では,ANGPTL2によるがん発症やがん病態の促進および抑制の作用機序,最近明らかとなった免疫チェックポイント阻害剤による免疫関連有害事象との関連についてもあわせて概説する.
ウイルスや細菌感染,化学物質への曝露等による慢性炎症は,胃がん,大腸がん,肝臓がん,皮膚がんなどさまざまながんの発症に関わっている23, 24).ヒトの皮膚組織では,非露光部に比べ露光部におけるANGPTL2の発現レベルが高く,加齢によってもその発現が増加することが明らかとなった25).加齢や紫外線など,がん発症のリスク因子によってANGPTL2発現が増加することから,ANGPTL2による慢性炎症が,皮膚がんの発症に寄与する可能性が示唆された.そこで,マウスの皮膚に変異誘発物質である7,12-dimethylbenzanthracene(DMBA)を単回塗布することでRas遺伝子に変異を導入し,その後,炎症誘導剤であるphorbol 12-myristate 13-acetate(PMA)を複数回塗布することで,扁平上皮がんの発生を誘導する多段階皮膚化学発がんモデル26)を用い,ANGPTL2と発がんとの関連を検討した.マウス正常皮膚組織に比べ,PMA塗布後の皮膚組織や,パピローマおよび扁平上皮がん組織において,Angptl2発現の増加が認められた25).発がんにおける皮膚組織でのAngptl2発現誘導の意義を検討するため,皮膚組織でANGPTL2を高発現するトランスジェニック(K14-Angptl2 Tg)マウスを作製したところ,皮膚組織におけるinterleukin-6(Il-6)やinterleukin-1β(Il-1β)などの炎症関連遺伝子の発現上昇や活性酸素種(reactive oxygen species:ROS)の産生増加を認め,慢性炎症が惹起されていることが確認された25).さらに,K14-Angptl2 Tgマウスを用いて多段階皮膚化学発がんモデルを作製すると,野生型マウスに比べ扁平上皮がんの発症率が著しく増加した25).逆に,Angptl2 KOマウスの皮膚組織では,野生型マウスに比べ炎症関連遺伝子の発現誘導やROS産生が軽減しており,扁平上皮がんの発症も抑制された25).以上より,皮膚組織でのANGPTL2過剰産生は,慢性炎症を惹起し,発がんの感受性を高めることが明らかとなった4, 25).
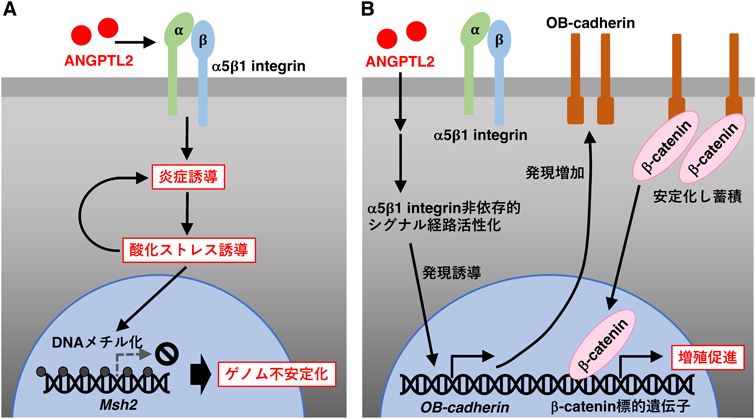
慢性炎症とこれに伴う酸化ストレスは,DNA損傷を引き起こし,変異蓄積によるゲノム不安定化など,発がんのリスクを高めることが知られている27, 28).K14-Angptl2 Tgマウスでは,ROS産生が増加していたことから,抗酸化剤であるN-acetylcysteine(NAC)を投与したところ,皮膚組織における酸化ストレスに加え,炎症関連遺伝子の発現レベルも低下し,扁平上皮がんの発症が抑制されることが明らかとなった25, 29).また,多段階皮膚化学発がんモデルでは,発がん誘導に伴い皮膚組織におけるDNAミスマッチ修復の鍵因子であるMutS homolog 2(Msh2)の発現が低下すること,K14-Angptl2 Tgマウスの皮膚組織では,野生型マウスに比べ,Msh2発現低下が促進されることが明らかとなった29).その後の解析から,K14-Angptl2 Tgマウスの皮膚組織では,Msh2プロモーター領域のDNAメチル化が亢進しており,NAC投与によってDNAメチル化および発現低下が抑制されることが明らかとなった29).以上より,皮膚組織におけるANGPTL2の過剰産生は,炎症だけでなく酸化ストレスを誘導することで,炎症の遷延化に加え,プロモーター領域のDNAメチル化促進によってMsh2発現を抑制し,DNA修復不全によるゲノム不安定化とこれによる発がんの感受性亢進に寄与することが示唆された4, 25, 29)(図3A).
大腸がん発症においてもANGPTL2シグナルとの関連が明らかとなっている.ヒト大腸がんの多くは,adenomatous polyposis coli(APC)やβ-cateninを含むWnt–β-catenin経路の構成分子の変異が発がんに関わる13, 30).通常,Wnt–β-catenin経路の古典的制御機構では,Wntシグナルがない場合,β-cateninタンパク質は,APCを含むβ-カテニン分解複合体によってリン酸化され,その後,ユビキチンプロテアソーム経路で分解される.しかし,変異型β-cateninタンパク質は,同複合体依存的な分解に抵抗性を示すことで持続的に蓄積し,結果として細胞増殖が促進され,がん発症に至る13, 30).変異誘発物質であるアゾキシメタン(azoxymethane:AOM)を頻回腹腔内投与することで大腸がん発症を誘発するAOM誘発大腸がんマウスモデルでは,腸管上皮細胞においてβ-catenin遺伝子に高頻度に変異が生じる31).我々は,同マウスモデルにおいて,非腫瘍部位の腸管上皮細胞ではANGPTL2の発現を認めないが,がん細胞ではANGPTL2が高発現していることを明らかにした32).そこで,Angptl2 KOマウスを用いてAOM誘発大腸がんマウスモデルを作製したところ,野生型マウスに比べ,大腸がんの発症が抑制されることが明らかとなった32).また,野生型マウスに比べ数は少ないものの,Angptl2 KOマウスに生じた大腸がん組織を解析したところ,β-cateninタンパク質の蓄積が軽減されており,β-cateninの標的遺伝子であるc-Mycやcyclin Dなどの細胞増殖関連遺伝子の発現が減少していた32).以上より,がん細胞由来のANGPTL2は,β-cateninタンパク質の蓄積を促進することで,がん細胞の増殖能を亢進し,大腸がん発症に寄与することが示唆された.
ANGPTL2によるβ-cateninタンパク質の蓄積促進メカニズムを検討したところ,ANGPTL2がα5β1 integrin非依存的にcadherin 11(CDH11,別名OB-cadherin)の発現を誘導すること,OB-cadherinの発現増加に伴いOB-cadherinとβ-cateninの相互作用が亢進することで,β-cateninタンパク質がさらに安定化し,その蓄積が促進されることが明らかとなった32)(図3B).以上より,新たな大腸がん発症機構として,従来の古典的制御機構とは独立したANGPTL2–OB-cadherin経路によるβ-cateninタンパク質の安定化機構が,腫瘍細胞の増殖促進に寄与しており,今後,同機構が新たな治療標的となることも期待される.
1)転移とANGPTL2
原発巣では,がん細胞の増殖に伴い,低栄養や低酸素といった環境の変化が生じる33, 34).また,がん組織内には,がん細胞以外にも血管細胞,線維芽細胞,免疫細胞などのさまざまな間質細胞が存在し,がん細胞と間質細胞間の相互作用が,がん進展に寄与する33, 35, 36).がん細胞は,このようながん微小環境の変化への応答により,その形質を変化させる.たとえば,がん微小環境の変化により,がん細胞では,上皮細胞から間葉系細胞へ形質が変化する上皮間葉転換(epithelial-mesenchymal transition:EMT)が生じ,遊走能や浸潤能の亢進,治療抵抗性の獲得など,浸潤・転移が促進される37, 38).転移能の亢進により血管内へと侵入したがん細胞は,血流を介して遠隔臓器へと移動し,血管外へ漏出することで転移巣が形成される37, 38).
我々を含め多くの研究グループが,がん組織におけるANGPTL2発現量や血中ANGPTL2濃度が,がん病態の進展や予後と連関することを報告している.我々は,非小細胞肺がん患者の原発巣組織におけるANGPTL2の発現量が高いグループと低いグループでは,ANGPTL2発現量が高いグループの無病生存期間および全生存期間が有意に短いことを明らかにした39).また,乳がん患者では,非浸潤性乳管がんや転移を認めない浸潤性乳管がんに比べ,転移を認める浸潤性乳管がんにおいて,血中ANGPTL2濃度が有意に高いことを明らかにした40).
上述の多段階皮膚化学発がんモデルを用いた研究において,K14-Angptl2 Tgマウスでは,発がんが促進されるだけでなく,肺やリンパ節への転移が増加し,野生型マウスに比べ,生存期間も著しく短縮する25).一方で,Angptl2 KOマウスでは,発がんの頻度も低いが,扁平上皮がんを発症しても,肺やリンパ節への転移が抑制され,生存期間の延伸が認められた25).また,ANGPTL2の発現量が異なるヒト肺がんや骨肉腫の細胞株をそれぞれ用いた異所性もしくは同所性の異種移植モデルでは,いずれのがんにおいても,ANGPTL2発現レベルが低いがん細胞に比べ,ANGPTL2発現レベルが高いがん細胞では,肺やリンパ節への転移が増加し,モデルマウスの生存期間が短縮することが明らかとなった12, 39).さらに,ANGPTL2を高発現し,高い転移能を示すヒト乳がん,肺がんおよび骨肉腫細胞株を用いた異種移植モデルでは,がん細胞においてANGPTL2をノックダウン(KD)した場合,肺やリンパ節への転移が抑制され,生存期間の延伸を認めた12, 39).これらの結果から,がん細胞におけるANGPTL2の発現量と転移能が連関することが明らかとなり,ANGPTL2シグナル抑制が,がん転移に対する治療戦略となる可能性が示唆された.
2)腫瘍血管新生とANGPTL2
多段階皮膚化学発がんモデルでは,K14-Angptl2 Tgマウスのがん組織内において腫瘍血管新生およびリンパ管新生が亢進しており,逆にAngptl2 KOマウスでは,腫瘍血管新生およびリンパ管新生ともに,野生型マウスに比べ減少していた25).また同様に,ANGPTL2を高発現し,高い転移能を示すヒト乳がんおよび骨肉腫細胞株を用いた異種移植モデルでは,がん細胞におけるANGPTL2のKDにより,がん組織内における腫瘍血管新生が減少した12, 39).以上より,がん細胞由来ANGPTL2は,腫瘍血管新生およびリンパ管新生を促進することが明らかとなった.腫瘍血管は,正常血管と比較して透過性が亢進しているため,がん細胞への栄養の供給だけでなく,がん転移にも寄与している37, 38).このことから,腫瘍血管新生の阻害はがん治療戦略として期待され,血管新生因子であるvascular endothelial growth factor(VEGF)に対する中和抗体を用いた腫瘍血管新生阻害が,複数のがんに対する治療法として知られている41).VEGFを同定し,抗VEGF中和抗体を開発したFerraraらのグループは,抗VEGF抗体治療に対して抵抗性を示すがんでは,がん組織内にANGPTL2を含む血管新生促進分子を高発現する線維芽細胞が存在し,腫瘍血管新生を促進することを報告している42).このことは,腫瘍微小環境において,がん細胞以外の間質細胞に由来するANGPTL2も,腫瘍血管新生を介してがん進展に寄与していることを示唆している.
3)EMTとANGPTL2
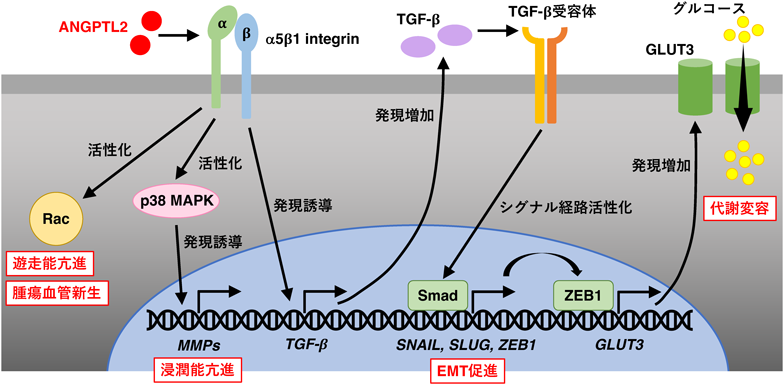
EMTの誘導には,さまざまなシグナル経路が関連しており,TGF-βシグナルはEMT関連転写因子の発現を活性化することでEMTを誘導する43).多段階皮膚化学発がんモデルにおいて,K14-Angptl2 Tgマウスでは,野生型マウスに比べ,がん組織におけるTgf-βやその受容体の発現およびSmad2のリン酸化が亢進していた25).さらに,TGF-βシグナル経路活性化によって発現誘導されるEMT関連転写因子であるSnailやSlugの発現も増加していた.また,上皮細胞マーカーであるE-cadherinの発現減少と間葉系細胞マーカーであるN-cadherinの発現増加が認められ,EMTが促進されていることが確認された.逆に,Angptl2 KOマウスでは,EMTの誘導は抑制されていた.さらに,ANGPTL2を高発現し,高い転移能を示すヒト肺がん細胞株であるLNM35においてANGPTL2をKDすると,TGF-βシグナルの減弱,SNAILおよびSLUGの発現減少とE-cadherin発現の増加が認められた39).逆に,LNM35細胞に比べANGPTL2発現レベルおよび転移能が低いヒト肺がん細胞株であるH460においてANGPTL2を過剰発現すると,TGF-βシグナル活性化やSNAILおよびSLUGの発現増加,E-cadherinの発現減少を認めた39).また,ANGPTL2を過剰発現したH460細胞におけるTGF-βの発現誘導は,α5β1 integrinに対する中和抗体によって抑制されることから,がん細胞由来ANGPTL2は,α5β1 integrinを介してパラクリン的あるいはオートクリン的に作用し,TGF-βの発現を誘導することで,EMTを促進することが明らかとなった39)(図4).我々は,慢性腎臓病の病態において,ANGPTL2–α5β1 integrin経路が,ERK活性化を介して尿細管上皮細胞におけるTGF-βの発現を誘導することを解明しているが22),がん病態におけるANGPTL2–α5β1 integrin経路を介したTGF-βの発現誘導機構の詳細は解明されていない.
4)がん細胞の遊走・浸潤とANGPTL2
がん細胞におけるANGPTL2の過剰産生は,組織リモデリングに関わるANGPTL2シグナル経路の過剰応答により,がん細胞の遊走能や浸潤能を高めることが明らかとなっている.上述のANGPTL2発現量が異なるヒト肺がん細胞株H460(低発現)とLNM35(高発現)を比較すると,LNM35細胞では活発に葉状仮足が形成され,ANGPTL2をKDすることで,葉状仮足形成が抑制された39).さらに,ANGPTL2を過剰発現したH460細胞では,葉状仮足の形成および細胞の遊走能が亢進し,葉状仮足形成に関わるRacの活性が増加することが明らかとなった39).また,ANGPTL2を高発現し,高い転移能を示すヒト骨肉腫細胞株143Bでは,ANGPTL2のKDやα5β1 integrinに対する中和抗体によってp38 MAPKの活性や,細胞外マトリックスのリモデリングに関わるMMP-9やMMP-13などの発現が低下すること,p38 MAPK活性阻害によってMMP活性が減弱し,その結果,浸潤能の低下につながることが明らかとなった12).以上より,がん細胞由来ANGPTL2は,Rac活性化およびp38 MAPK–MMP経路活性化によりがん細胞の遊走能や浸潤能を亢進し,転移を促進することが示唆された(図4).143B細胞を用いた同所性の異種移植モデルでは,がん組織内の腫瘍血管新生の増加に加え,腫瘍血管内腔に多数のがん細胞が侵入しており,がん細胞においてANGPTL2をKDすると血管内侵入が抑制された12).一方で,ANGPTL2をKDした143B細胞を尾静脈投与し,肺における転移巣の形成を評価したところ,ANGPTL2をKDしていない143B細胞と比べても差は認められなかった12).これらの結果は,ANGPTL2によるがん細胞の転移能の活性化が,原発巣における血管内侵入を促進するが,転移先の遠隔臓器における血管外漏出には寄与しないことを示唆している.
乳がんでは,リンパ節,肺,肝臓,脳,骨が,遠隔転移しやすい臓器として知られている.このような乳がんの転移先の臓器選択性にANGPTL2が関与していることが明らかとなった.我々は,ANGPTL2を高発現し,高い転移能を示すヒト乳がん細胞株MDA-MB-231においてANGPTL2をKDすると,ケモカイン受容体であるC-X-C motif chemokine receptor type 4(CXCR4)の発現が減少することを見いだした44).ANGPTL2によるCXCR4の発現誘導機構を解析したところ,ANGPTL2シグナルによって転写因子ETS1の発現が誘導され,ETS1によってCXCR4の転写が活性化されることが明らかとなった44).CXCR4のリガンドは,C-X-C motif chemokine ligand 12(CXCL12)であり,骨を含むさまざまな臓器で発現している45, 46).上述のように,ANGPTL2シグナルは原発巣におけるがん転移過程に影響するため,コントロールまたはANGPTL2をKDしたMDA-MB-231細胞を免疫不全マウスの左心室内に投与することで,転移巣形成能だけを評価したところ,ANGPTL2をKDしたMDA-MB-231細胞を投与した場合に,骨転移が抑制され,生存期間の延伸が認められた44).さらに,ANGPTL2をKDしたMDA-MB-231細胞では,CXCL12–CXCR4経路によるERK活性化およびMMP-13の発現が減弱し,骨基質の大部分を占めるI型コラーゲンに対する浸潤が減少した44).また,CXCL12–CXCR4経路による乳がん細胞の浸潤促進作用は,ERK活性阻害によって抑制された44).以上より,乳がん細胞では,ANGPTL2–ETS1経路によってCXCR4の発現が増加し,CXCL12が産生される骨組織への誘引が促進されるとともに,CXCL12–CXCR4経路によるMMP-13の発現および活性の亢進により,骨基質の分解による組織内への浸潤と生着が促進されることが示唆された.
5)がん細胞代謝とANGPTL2
がん細胞は,有酸素環境下においても,ミトコンドリアによる酸化的リン酸化よりも解糖系を利用してATPを産生する.Warburg効果として知られるこの現象は,がん微小環境が低酸素状態にあっても解糖系で生存のためのエネルギーを得るために獲得する形質であると考えられてきたが,近年では,解糖系を介して核酸やnicotinamide adenine dinucleotide phosphate(NADPH)などの生体分子の材料を得るためであるとする説が有力視されている.我々は,ANGPTL2が,がん細胞におけるこのような代謝変容に関わっていることを明らかにした.ANGPTL2発現レベルおよび転移能が低いヒト肺がん細胞株H460に比べ,ANGPTL2を高発現し,高い転移能を示すヒト肺がん細胞株LNM35では,細胞内へのグルコースの取り込み能が高く,乳酸産生も多いことから,がん細胞におけるANGPTL2発現量と解糖系亢進との連関が示唆された47).さらに,LNM35細胞では,グルコース輸送体であるglucose transporter 3(GLUT3)の発現が亢進しており,ANGPTL2のKDによってその発現が抑制された47).上述のように,肺がん細胞において,ANGPTL2は,α5β1 integrinを介してTGF-βシグナル経路活性化とこれに伴うEMT関連転写因子であるSNAILやSLUGの発現を誘導するが,同じくEMT関連転写因子であるZEB1の発現を誘導し,ZEB1がGULT3の転写を活性化することが明らかとなった43, 47)(図4).肺がん細胞では,ANGPTL2–α5β1 integrin経路活性化によって,EMT誘導と代謝変容が連動することで,転移能を高めいている可能性が考えられる.
6)治療抵抗性とANGPTL2
がん治療においては,薬物療法に対する治療抵抗性の出現が大きな問題となる.がん細胞における治療抵抗性獲得のさまざまなメカニズムが報告されている.上述のように,がん組織内の線維芽細胞におけるANGPTL2発現は,腫瘍血管新生を標的とした抗VEGF抗体治療の治療抵抗性に関連する42).さらに,5-fluorouracil(5-FU)は,さまざまながんの化学療法で用いられ,DNA合成を阻害することでがん細胞のアポトーシスを誘導するが,我々は,大腸がんにおいて,ANGPTL2シグナルが5-FUによるアポトーシスを抑制し,抵抗性をもたらすことを解明した48).そのメカニズムとして,ANGPTL2は,ヒト大腸がん細胞におけるspleen tyrosine kinase(SYK)の発現および活性化を促進することで,phosphatidylinositol 3-kinase(PI3K)の活性化とこれに伴うNF-κBの核内移行を引き起こし,B-cell/CLL lymphoma 2(BCL2)やBCL-XLといったアポトーシス抑制に作用するBCL2ファミリー分子の発現を誘導することが明らかとなった48).
がん幹細胞は,高い自己複製能,造腫瘍性,転移能,薬剤耐性を有し,治療抵抗性獲得,再発および転移にも寄与する49, 50).固形腫瘍におけるANGPTL2とがん幹細胞との関連についてはこれまで報告はないが,造血器腫瘍においては,がん幹細胞の維持にANGPTL2が寄与することが報告されている.急性骨髄性白血病(acute myeloid leukemia:AML)は,骨髄系幹細胞を起源として発生し,未成熟な白血病細胞が異常増殖することで,正常な血液細胞の産生を阻害する.造血幹細胞では,LILRB2/PIR-BがANGPTL2受容体として機能しているが,ヒト白血病細胞においてもLILRB2/PIR-Bが発現している17).Zhangらのグループは,AMLの原因遺伝子であるMLL-AF9やAML1-ETO9αを導入した未成熟の骨髄細胞をマウスに移植することでAMLを発症する白血病マウスモデルを用いた研究から,野生型のドナー細胞に比べ,Pir-BをKOしたドナー細胞を移植すると,白血病細胞数の減少やモデルマウスの予後が改善することを示した17).また,ANGPTL2が,造血幹細胞の維持に関わるLILRB2/PIR-B–CAMK4経路を活性化し,白血病の発症・進展および白血病幹細胞の幹細胞性維持を促進することも報告されている17).さらに,最近,骨髄内の血管内皮細胞に由来する細胞外小胞にANGPTL2が豊富に含まれており,詳細なメカニズムは不明だが,これがLILRB2経路を活性化し,白血病の発症・進展および白血病幹細胞の維持に寄与していることも報告されている51).
がん細胞におけるANGPTL2の発現量と転移能が連関することから,がん細胞におけるANGPTL2発現制御機構解明は重要である.我々は,上述のANGPTL2を高発現し,高い転移能を示すヒト骨肉腫細胞株143BよりもANGPTL2発現レベルが低いヒト骨肉腫細胞株SaOS2を用いた同所性の異種移植モデルの解析過程において,原発巣のがん細胞におけるANGPTL2発現量が,移植後,経時的に増加することを見いだした12).そこで,ANGPTL2プロモーター領域におけるDNAメチル化レベルを解析したところ,移植後,DNAメチル化レベルが低下することが明らかとなった12).ANGPTL2発現量が異なる乳がん,膵がん,骨肉腫などのさまざまな種類のヒトがん細胞株においても,ANGPTL2発現量とプロモーター領域のDNAメチル化レベルは逆相関した12).また,SaOS2細胞の異種移植モデルにおいて,肺転移したがん細胞を単離し,解析したところ,移植前のSaOS2細胞に比べ,ANGPTL2プロモーター領域のDNAメチル化レベルは低下しており,その発現は亢進したままの状態となっていた12).これらの結果から,原発巣のがん微小環境が,がん細胞におけるANGPTL2プロモーター領域のDNA脱メチル化を促進することで,ANGPTL2発現が亢進し,結果として転移能が活性化したがん細胞が,遠隔臓器に転移すると考えられた.
我々は,移植したSaOS2細胞において,DNA脱メチル化酵素であるten-eleven translocation(TET)ファミリー52)のなかでも,TET2の発現が亢進していること,がん細胞を低酸素・低栄養環境下で培養すると,ANGPTL2およびTET2の発現が誘導されることを明らかにした12, 53).さらに,TET2をKDしたSaOS2細胞を用い,同所性の異種移植モデルを作製したところ,原発巣におけるANGPTL2発現誘導が抑制された.また,コトントロールと比べてもTET2をKDしたSaOS2細胞を移植したマウスの腫瘍の成長には差を認めないが,肺転移は有意に減少した53).以上より,原発巣でのがん細胞の増殖に伴う低酸素や低栄養といったがん微小環境の変化が,がん細胞におけるTET2発現を誘導し,ANGPTL2プロモーター領域のDNA脱メチル化が促進されることで,転移促進につながることが示唆された.
さらに,我々は,ANGPTL2発現誘導に関わる転写制御因子として,nuclear factor activated T cell 2(NFATc2),activating transcription factor 2(ATF2),およびc-Junが重要であることを解明した39).ヒトANGPTL2プロモーター領域には,ATF/cAMP-responsive element(CREB)結合配列が存在しており,ここにATF2とc-Junの複合体が直接結合すること,NFATc2はATF2とc-Junの複合体を介して結合し,NFATc2を含む複合体がANGPTL2の転写を強力に活性化することが明らかとなった39).NFATは細胞内のカルシウム濃度上昇によって活性化する転写因子であり,ATF2とc-Junもさまざまなストレスによって活性化することが知られている54–56).がん細胞の増殖に伴う低酸素や低栄養は,小胞体ストレスを誘導するなど,がん細胞の細胞内カルシウム濃度変化やストレス経路活性化に寄与し,NFATc2–ATF2–c-Jun複合体の活性化を引き起こすことで,ANGPTL2発現が誘導されると考えられる.
我々は,上述のようながん微小環境の変化によるANGPTL2の発現誘導機構に加え,最近,がんの原因遺伝子による直接のANGPTL2発現誘導機構を解明した.Xp11.2転座型腎細胞がん(translocation renal cell carcinoma:tRCC)は,小児や若年者に発症する予後不良な希少腎がんであり,その原因遺伝子としてXp11.2転座により生じる転写因子transcription factor E3(TFE3)の融合遺伝子が複数同定されている57).本来,TFE3は栄養飢餓などのストレスに応答して活性化することで核内へと移行し,細胞代謝やオートファジー制御関連遺伝子の発現を活性化する58).融合した遺伝子にかかわらず,TFE3融合遺伝子の翻訳産物であるTFE3キメラタンパク質は,そのC末端側にDNA結合ドメインを有しており,ストレス非依存的に核内へ移行し,持続的にTFE3標的遺伝子発現を誘導することで病態発症につながると考えられている57).我々は,ヒト尿細管上皮細胞において,PRCC-TFE3, NONO-TFE3,およびSFPQ-TFE3の3種類のTFE3キメラタンパク質によって発現誘導される遺伝子群を解析したところ,各TFE3キメラタンパク質によって共通して発現誘導される遺伝子の一つがANGPTL2であることを解明した59).また,TFE3キメラタンパク質によるANGPTL2の発現誘導機構として,TFE3キメラタンパク質が,ANGPTL2プロモーター領域に存在する2か所のE-box配列に結合し,転写を活性化することを解明した60).ANGPTL2プロモーター領域に存在するE-box配列は,前述のNFATc2–ATF2–c-Jun複合体が結合するATF/CREB結合配列から約1.2 kbほど上流に存在している.TFE3キメラタンパク質とNFATc2–ATF2–c-Jun複合体は協調してANGPTL2の転写を活性化する可能性も考えられるが,詳細については解明されていない.
1)間質細胞由来ANGPTL2によるがん抑制作用
TFE3キメラタンパク質によってANGPTL2の発現が誘導されることから,tRCCの発症・進展にもANGPTL2が寄与していることが示唆された.そこで我々は,尿細管上皮細胞特異的にPRCC-TFE3を高発現することで,腎細胞がんを発症するtRCCマウスモデルを開発し,がん病変においてANGPTL2の発現が亢進していることを確認した59, 61).tRCCの発症・進展とANGPTL2との関連を検討するため,尿細管上皮細胞特異的にAngptl2をKOしたtRCC[conditional KO(CKO)tRCC]モデルマウスを作製したところ,予想どおり,腎細胞がんの発症・進展が遅延し,野生型のtRCCモデルマウスに比べ生存期間が延伸した59, 60).ところが,全身でAngptl2をKOしたtRCC[systemic KO(SKO)tRCC]モデルマウスを作製したところ,野生型のtRCCモデルマウスに比べ,腎がん病態が増悪し,その生存期間も短縮した59).これらの結果から,尿細管上皮細胞またはがん細胞由来のANGPTL2は,がん病態促進に寄与するが,間質由来のANGPTL2は,がん病態抑制に作用することが考えられた.実際,野生型およびAngptl2 KOマウスの皮下に,ANGPTL2を発現していないことが確認されたマウスメラノーマ細胞株B16F10を移植すると,Angptl2 KOマウスにおいて腫瘍の成長が促進され,生存期間も短縮した59).以上より,間質由来ANGPTL2は,がん抑制作用を有することが明らかとなった.
tRCCモデルマウスやB16F10細胞を移植したマウスのがん組織を用い,間質においてANGPTL2を発現する細胞を探索したところ,platelet-derived growth factor receptor α(PDGFRα)を発現するがん関連線維芽細胞(cancer-associated fibroblast:CAF)がANGPTL2を発現していることが明らかとなった59).さらに,Angptl2 KOマウスに野生型マウス由来PDGFRα陽性CAFとがん細胞を同時に移植すると,腫瘍の成長が抑制され,生存期間も延伸した59).また,SKO tRCCモデルマウスやB16F10細胞を移植したAngptl2 KOマウスのがん組織では,CD8+ T細胞の浸潤が減少していることを見いだした59).モデルがん抗原としてovalbumin(OVA)を発現するB16F10細胞を移植し,がん特異的な免疫応答を評価したところ,Angptl2 KOマウスでは,OVAに対して特異的に応答するCD8+ T細胞が減少しており,がん抗原特異的CD8+ T細胞の誘導が減弱していた59).以上より,PDGFRα陽性CAF由来ANGPTL2は,抗腫瘍免疫応答を促進することが示唆された.
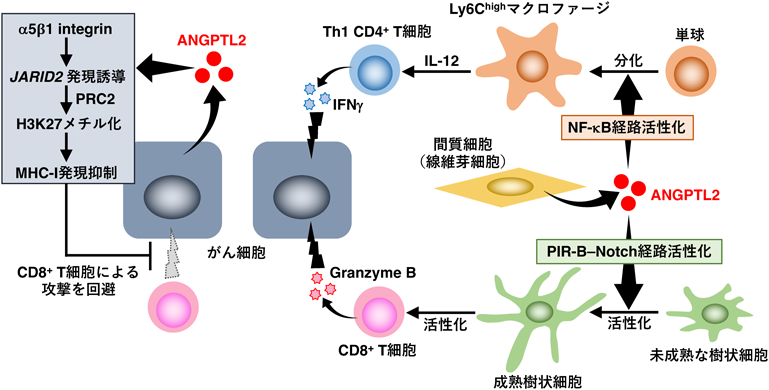
Angptl2 KOマウスでは,がん抗原特異的CD8+ T細胞の誘導が減弱していたことから,ANGPTL2は,樹状細胞によるCD8+ T細胞の活性化(cross-priming)に寄与していることが考えられた.我々は,樹状細胞に対するANGPTL2の作用を詳細に検討したところ,ANGPTL2が,α5β1 integrinではなくPIR-Bを介してNotch2の活性化を促進することで,CD8+ T細胞の活性化に重要なIL-12や共刺激分子(CD86, CD80, CD40)および抗原提示に重要なmajor histocompatibility complex(MHC)class IIの発現を亢進するなど,樹状細胞の成熟および活性化を促進し,CD8+ T細胞による抗腫瘍免疫応答を活性化していることを解明した59)(図5).
間質細胞由来ANGPTL2による抗腫瘍免疫応答の活性化は,tRCCマウスモデルのみならず,大腸炎関連発がんマウスモデルにおいても認められた.上述のAOM誘発大腸がんマウスモデルとは異なり,大腸炎関連発がんマウスモデルでは,AOMを腹腔内に単回投与した後,DSS飲水投与5日間に続く16日間の休止期間を1サイクルとし,これを3サイクル行うことで大腸がんの発症を誘導する62).AOM誘発大腸がんマウスモデルは,発がんに炎症を伴わないが,大腸炎関連発がんマウスモデルでは,DSSによる組織損傷によって炎症が誘導され,発がんを促進する.Angptl2 KOマウスを用いて大腸炎関連発がんマウスモデルを作製したところ,野生型マウスに比べ,腫瘍の成長が促進され,生存期間が短縮することが明らかとなった63).また,がん組織内では,がん細胞に加え,筋線維芽細胞がANGPTL2を豊富に発現しており,筋線維芽細胞由来ANGPTL2が,がん抑制作用を示すことが考えられた.実際,Angptl2 KOマウスでは,がん組織や腸炎を発症した腸管組織内に浸潤したT細胞,マクロファージ,好中球などが減少していた.特に,T細胞では,interferon γ(IFNγ)を発現するCD4+ T細胞が減少し,マクロファージでは,Ly6Cを高発現(Ly6Chigh)する骨髄由来マクロファージが減少していた63).ANGPTL2とこれらの免疫細胞との関連を検討したところ,ANGPTL2は,マクロファージにおいてNF-κBを活性化することで,Ly6Chighマクロファージへの分化を促進することが明らかとなった63).また,CD4+ T細胞には,その機能の違いから,複数のサブセットが存在し,Th1タイプに分類される上記CD4+ T細胞は,IFNγを産生することで,直接がん細胞を障害するだけでなく,がんに対する細胞性免疫の活性化にも寄与する64, 65).Ly6Chighマクロファージは,抗原感作を受けていないナイーブCD4+ T細胞をTh1タイプに分化させるIL-12を発現しており,ANGPTL2はマクロファージにおけるIL-12産生を亢進し,ナイーブCD4+ T細胞からTh1 CD4+ T細胞への分化を促進することが明らかとなった63).以上より,腸管組織の筋線維芽細胞由来ANGPTL2は,NF-κB経路活性化を介して免疫賦活性の表現型を示すマクロファージへの分化を促進し,Th1 CD4+ T細胞による抗腫瘍免疫応答を活性化することが示唆された(図5).
2)がん細胞由来ANGPTL2による免疫逃避機構
細胞死を起こしたがん細胞やがん細胞由来の変異遺伝子産物を貪食した抗原提示細胞は,MHC class I(MHC-I)を介してがん細胞由来のペプチド(がん抗原)を提示することで,がん特異的CD8+ T細胞の活性化・増殖を引き起こす66, 67).がん細胞表面にも細胞内で分解されたがん抗原ペプチドがMHC-Iを介して提示されており,がん特異的CD8+ T細胞はこれを認識し,がん細胞を排除する66, 67).一方で,がん細胞はこのような抗腫瘍免疫から逃れる免疫逃避を行うことが知られている68, 69).その機序の一つとして,一部のがん細胞では,MHC-I遺伝子の欠失が認められる場合もあるが,同遺伝子の欠失がない場合でもその発現を低下させることで,CD8+ T細胞による認識を回避するという免疫逃避機構が存在している68, 69).しかし,がん細胞におけるMHC-I発現抑制の詳細な分子機構は十分に解明されていなかった.上述の尿細管上皮細胞でAngptl2をKOしたCKO tRCCモデルマウスでは,これまでの我々の研究成果と一致して,がん細胞由来ANGPTL2が,がん病態進展に寄与することが確認された59, 60).これまでに紹介したANGPTL2–α5β1 integrin経路を介したがん細胞の運動能や浸潤能,EMTなどの促進が,tRCCの病態進展にも寄与していると考えられたが,間質由来ANGPTL2の抗腫瘍免疫活性化によるがん抑制作用が明らかとなったことから,がん細胞由来ANGPTL2が,抗腫瘍免疫応答を抑制する可能性を検証した.その結果,CKO tRCCモデルマウスの腎組織では,野生型tRCCモデルマウスに比べ,細胞障害活性分子であるgranzyme Bを発現する活性化CD8+ T細胞が増加していることを見いだした60).さらに,我々は,野生型tRCCモデルマウスのがん組織より樹立した初代がん細胞株(tRCC細胞)を用い,Angptl2をKOしたところ,Angptl2 KO tRCC細胞ではMHC-Iの発現が亢進していることが明らかとなった60).さらに,MHC-Iや細胞内でのがん抗原分解に関わる抗原提示関連分子の発現は,IFNγによって誘導されることが知られており68, 70, 71),Angptl2 KO tRCC細胞では,IFNγ誘導性のMHC-I発現が促進された60).また,受容体であるα5β1 integrinのうち,α5 integrinをKOしたtRCC細胞においても同様の結果を得た.これらの結果より,がん細胞由来ANGPTL2は,α5β1 integrinを介してMHC-I発現を抑制することで,がん細胞の免疫逃避に寄与していることが示唆された.そこで,モデルがん抗原としてOVAを発現するAngptl2 KO tRCC細胞を作製し,同細胞に対するOVA特異的なT細胞受容体を発現するCD8+ T細胞(OT-I細胞)の障害活性を評価した.その結果,野生型tRCC細胞に比べ,Angptl2 KO tRCC細胞では,IFNγ刺激を行った際の細胞表面におけるMHC-I–OVAペプチド複合体の発現が亢進し,OT-I細胞による細胞障害活性が増加することが明らかとなった60).以上より,ANGPTL2は,がん細胞におけるIFNγ誘導性のMHC-I発現を阻害し,がん抗原特異的なCD8+ T細胞の活性化とこれによるがん細胞の排除を抑制していることが示された.
IFNγシグナルは,転写因子signal transducer and activator of transcription 1(STAT1)を活性化することで,その一次応答遺伝子であるinterferon regulatory factor 1(Irf1)やNLR family CARD domain containing 5(Nlrc5)の発現を誘導する70, 71).IRF1とNLRC5は,さまざまな因子と転写活性化複合体を形成することで,MHC-Iをはじめとする標的遺伝子の発現を誘導する70, 71).がん細胞では,IFNγシグナル経路関連分子の欠失によって応答不全となることが知られているが,野生型tRCC細胞とAngptl2 KO tRCC細胞では,IFNγ刺激後のSTAT1活性化やIrf1およびNlrc5の誘導に差を認めなかったことから,ANGPTL2はIFNγシグナル経路には影響しないことが明らかとなった60).
最近,がん細胞におけるMHC-Iを含む抗原提示関連分子の発現抑制メカニズムとして,エピゲノム制御との関連が報告された72).そこで,野生型tRCC細胞とAngptl2 KOまたはα5 integrin KO tRCC細胞のMHC-I遺伝子プロモーター領域のヒストン修飾を解析したところ,いずれのKO細胞においても,転写抑制型のヒストンH3リシン27トリメチル化(H3K27me3)が減少していることを見いだした60).H3K27は,polycomb repressive complex 2(PRC2)によってメチル化されることが知られている73).PRC2には,PRC2.1とPRC2.2と呼ばれるバリアントが存在し,いずれもメチル基転移酵素活性を有するenhancer of zeste homolog 2(EZH2)や,embryonic ectoderm development(EED)およびSUZ12といったコアサブユニットから構成されるが,各バリアントでコアサブユニット以外の構成因子が異なる73, 74).我々は,野生型tRCC細胞とAngptl2 KOまたはα5 integrin KO tRCC細胞では,コアサブユニットやPRC2.1の構成因子であるmetal response element binding transcription factor 2(Mtf2)の発現に差を認めないが,PRC2.2の構成因子であるjumonji and AT-rich interaction domain containing 2(Jarid2)の発現が,いずれのKO細胞においても減少していることを見いだした60).JARID2は,EZH2の酵素活性を亢進することや,標的遺伝子へのPRC2.2のリクルートを促進することが報告されている73, 74).実際,Angptl2 KOまたはα5 integrin KO tRCC細胞では,MHC-I遺伝子プロモーター領域におけるEZH2の結合量が減少しており,JARID2を過剰発現したAngptl2 KO tRCC細胞では,EZH2の結合量が回復し,MHC-Iの発現も抑制された60).以上より,ANGPTL2は,α5β1 integrinを介してがん細胞におけるJARID2の発現を誘導し,PRC2によるMHC-I遺伝子プロモーター領域の転写抑制型ヒストン修飾を促進することで,MHC-Iの発現および細胞表面へのがん抗原提示を抑制すること,これによりCD8+ T細胞による抗腫瘍免疫応答を回避することが解明された(図5).
がん細胞は,自己に対する免疫応答や,過剰な免疫応答を抑制する免疫チェックポイント機構を活性化し,抗腫瘍免疫応答を抑制する.近年,programed cell death protein-1(PD-1)やそのリガンドであるPD-1 ligand-1(PD-L1)などの免疫チェックポイント分子を標的とした免疫チェックポイント阻害剤が,さまざまな種類のがんに対する有効ながん免疫療法として臨床応用されている.しかしその一方で,免疫チェックポイント阻害剤で治療した患者の多くで,免疫関連有害事象と呼ばれる自己免疫疾患様の症状がさまざまな臓器で出現することが問題となっている75).心臓においては,免疫チェックポイント阻害剤による治療を受けた患者のうち,約1%が心筋炎を発症し,発症後の予後は不良とされている76).しかし,免疫関連有害事象の発現機構については,十分に解明されていない.
ANGPTL2は,樹状細胞やマクロファージによるT細胞免疫応答活性化を促進することから59, 63),心臓における免疫関連有害事象の発現にANGPTL2が関わっている可能性を考えた.そこで我々は,自己抗原としてαミオシン重鎖由来ペプチドを提示する樹状細胞をBALB/cマウスに移植することで,自己免疫性心筋炎を誘発する実験的自己免疫性心筋炎(experimental autoimmune myocarditis:EAM)モデルを樹立した77).このEAMモデルマウスにおいて,抗PD-1抗体および抗PD-L1抗体を同時に複数回腹腔内投与すると,心臓組織の炎症,樹状細胞,マクロファージ,CD4+およびCD8+ T細胞の浸潤,心機能低下が増悪することを確認した77).次に,EAMモデルマウスの心臓組織におけるANGPTL2発現細胞を検討したところ,特に心臓の筋線維芽細胞でANGPTL2が豊富に発現していることが明らかとなった77).また,血中ANGPTL2濃度は,心筋炎を誘導していないコントロールマウスに比べ,EAMモデルマウスで増加傾向にあり,免疫チェックポイント阻害剤投与により有意に増加した77).さらに,Angptl2 KOマウスを用いてEAMモデルを作製すると,野生型マウスで認められた免疫チェックポイント阻害剤による心筋炎および免疫細胞浸潤の増悪が改善され,心機能低下も抑制された77).
我々は,心臓の筋線維芽細胞がANGPTL2を豊富に発現する点に着目し,野生型およびAngptl2 KOマウスの筋線維芽細胞を解析したところ,Angptl2 KOマウス由来の細胞では,T細胞の浸潤を促進するさまざまなケモカインの発現が減少していることを見いだした77).さらに,ANGPTL2は,α5β1 integrinを介してNF-κB経路を活性化し,筋線維芽細胞におけるケモカインの発現を誘導していることを明らかにした77).以上の結果から,ANGPTL2は,心臓の筋線維芽細胞におけるα5β1 integrin–NF-κB経路を活性化することで,ケモカインの発現誘導とこれによるT細胞浸潤を促進し,免疫チェックポイント阻害剤による心筋炎の増悪に寄与していることが示唆された.
本稿で紹介したように,ANGPTL2は,がん病態促進だけでなく抑制にも作用するといった,がん発症・進展における二面性が明らかとなった.さまざまなヒトがん患者のがん組織における遺伝子発現と生存期間との連関について公共データベースを用いて解析すると,その多くは,ANGPTL2の発現レベルが低い患者群に比べ,ANGPTL2を高発現する患者群の予後が不良となり,ANGPTL2のがん促進作用を裏づける結果となった.しかし,一部では,逆にANGPTL2を高発現する患者群の方が予後良好となり,ヒトのがん病態においてもANGPTL2の二面性が認められた.さらに,がん病態進行の各ステージによって,ANGPTL2の作用が異なることも報告されている.ヒト卵巣がんでは,ステージI, IIといった早期であれば,がん組織におけるANGPTL2の発現が高い患者群の予後は良好であるが,ステージIII, IVと進行すると,逆に高発現群の予後は著しく不良となる78).しかし,がん病態に対する相反するANGPTL2の作用が,どのような機序で制御されているかはほとんど解明されていない.
がん病態に対するANGPTL2の作用制御に関しては,いくつかの機序が考えられる.一つ目として,免疫細胞の浸潤(炎症)の有無との関連である.同じ大腸がんであっても,免疫細胞の浸潤を伴わないAOM誘発大腸がんマウスモデルでは,ANGPTL2は発がんを促進したのに対し,免疫細胞の浸潤を伴う大腸炎関連発がんマウスモデルでは,ANGPTL2は発がん抑制に作用した.がん組織内で,ANGPTL2の標的細胞が主にがん細胞だけの場合,ANGPTL2はがん促進に作用するが,がん細胞以外にANGPTL2の標的となる樹状細胞やマクロファージなどの抗腫瘍免疫活性化に関わる免疫細胞が存在する場合,がん抑制に作用することが考えられる.二つ目として,標的細胞に発現している受容体の違いが寄与している可能性が考えられる.がん細胞ではα5β1 integrinが,樹状細胞ではLILRB2/PIR-Bが,それぞれANGPTL2受容体として機能するため,これらの細胞間でリガンドであるANGPTL2に対して競合が生じると予想される.この場合,それぞれの受容体を発現する細胞の数や受容体の発現量,各受容体に対するANGPTL2の親和性の違いが,がん病態に対するANGPTL2の作用を左右する要因になると考えられる.さらに,三つ目として,がん細胞と間質細胞におけるANGPTL2の翻訳後修飾の違いが関わっている可能性である.がん細胞と正常細胞では,糖鎖修飾構造が異なっており,がん細胞におけるさまざまな分子の糖鎖修飾の変容が,がん病態進展に関わっていることが報告されている79).ANGPTL2は,糖鎖修飾されることが明らかとなっており6),ANGPTL2を産生するがん細胞と間質細胞での糖鎖修飾構造の違いが,α5β1 integrinとLILRB2/PIR-B受容体への親和性に影響することも考えられる.今後,これらの可能性を検証し,がん病態におけるANGPTL2の作用制御機構を解明することが,ANGPTL2を標的としたがん治療戦略を開発する上で重要な課題であると考える.
謝辞Acknowledgments
本稿で紹介した研究は,熊本大学大学院生命科学研究部分子遺伝学講座に所属するスタッフや大学院生,多くの卒業生,ならびに学内外の共同研究者の皆様の協力のもとで行われたものであり,この場を借りて深く感謝申し上げます.
引用文献References
1) Medzhitov, R. (2008) Origin and physiological roles of inflammation. Nature, 454, 428–435.
2) Handschin, C. & Spiegelman, B.M. (2008) The role of exercise and PGC1alpha in inflammation and chronic disease. Nature, 454, 463–469.
3) Hato, T., Tabata, M., & Oike, Y. (2008) The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med., 18, 6–14.
4) Kadomatsu, T., Endo, M., Miyata, K., & Oike, Y. (2014) Diverse roles of ANGPTL2 in physiology and pathophysiology. Trends Endocrinol. Metab., 25, 245–254.
5) Kadomatsu, T. & Oike, Y. (2019) Roles of angiopoietin-like proteins in regulation of stem cell activity. J. Biochem., 165, 309–315.
6) Kim, I., Moon, S.O., Koh, K.N., Kim, H., Uhm, C.S., Kwak, H.J., Kim, N.G., & Koh, G.Y. (1999) Molecular cloning, expression, and characterization of angiopoietin-related protein. angiopoietin-related protein induces endothelial cell sprouting. J. Biol. Chem., 274, 26523–26528.
7) Oike, Y., Akao, M., Kubota, Y., & Suda, T. (2005) Angiopoietin-like proteins: Potential new targets for metabolic syndrome therapy. Trends Mol. Med., 11, 473–479.
8) Kubota, Y., Oike, Y., Satoh, S., Tabata, Y., Niikura, Y., Morisada, T., Akao, M., Urano, T., Ito, Y., Miyamoto, T., et al. (2005) Isolation and expression patterns of genes for three angiopoietin-like proteins, Angptl1, 2 and 6 in zebrafish. Gene Expr. Patterns, 5, 679–685.
9) Motokawa, I., Endo, M., Terada, K., Horiguchi, H., Miyata, K., Kadomatsu, T., Morinaga, J., Sugizaki, T., Ito, T., Araki, K., et al. (2016) Interstitial pneumonia induced by bleomycin treatment is exacerbated in Angptl2-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol., 311, L704–L713.
10) Horiguchi, H., Endo, M., Kawane, K., Kadomatsu, T., Terada, K., Morinaga, J., Araki, K., Miyata, K., & Oike, Y. (2017) ANGPTL2 expression in the intestinal stem cell niche controls epithelial regeneration and homeostasis. EMBO J., 36, 409–424.
11) Tabata, M., Kadomatsu, T., Fukuhara, S., Miyata, K., Ito, Y., Endo, M., Urano, T., Zhu, H.J., Tsukano, H., Tazume, H., et al. (2009) Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metab., 10, 178–188.
12) Odagiri, H., Kadomatsu, T., Endo, M., Masuda, T., Morioka, M.S., Fukuhara, S., Miyamoto, T., Kobayashi, E., Miyata, K., Aoi, J., et al. (2014) The secreted protein ANGPTL2 promotes metastasis of osteosarcoma cells through integrin alpha5beta1, p38 MAPK, and matrix metalloproteinases. Sci. Signal., 7, ra7.
13) Reya, T. & Clevers, H. (2005) Wnt signalling in stem cells and cancer. Nature, 434, 843–850.
14) He, X.C., Zhang, J., Tong, W.G., Tawfik, O., Ross, J., Scoville, D.H., Tian, Q., Zeng, X., He, X., Wiedemann, L.M., et al. (2004) BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet., 36, 1117–1121.
15) Powell, D.W., Pinchuk, I.V., Saada, J.I., Chen, X., & Mifflin, R.C. (2011) Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol., 73, 213–237.
16) Zhang, C.C., Kaba, M., Ge, G., Xie, K., Tong, W., Hug, C., & Lodish, H.F. (2006) Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat. Med., 12, 240–245.
17) Zheng, J., Umikawa, M., Cui, C., Li, J., Chen, X., Zhang, C., Huynh, H., Kang, X., Silvany, R., Wan, X., et al. (2012) Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature, 485, 656–660.
18) Lin, M.I., Price, E.N., Boatman, S., Hagedorn, E.J., Trompouki, E., Satishchandran, S., Carspecken, C.W., Uong, A., DiBiase, A., Yang, S., et al. (2015) Angiopoietin-like proteins stimulate HSPC development through interaction with notch receptor signaling. eLife, 4, e05544.
19) Sasaki, Y., Ohta, M., Desai, D., Figueiredo, J.L., Whelan, M.C., Sugano, T., Yamabi, M., Yano, W., Faits, T., Yabusaki, K., et al. (2015) Angiopoietin Like Protein 2 (ANGPTL2) Promotes Adipose Tissue Macrophage and T lymphocyte Accumulation and Leads to Insulin Resistance. PLoS One, 10, e0131176.
20) Horio, E., Kadomatsu, T., Miyata, K., Arai, Y., Hosokawa, K., Doi, Y., Ninomiya, T., Horiguchi, H., Endo, M., Tabata, M., et al. (2014) Role of endothelial cell-derived angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arterioscler. Thromb. Vasc. Biol., 34, 790–800.
21) Tazume, H., Miyata, K., Tian, Z., Endo, M., Horiguchi, H., Takahashi, O., Horio, E., Tsukano, H., Kadomatsu, T., Nakashima, Y., et al. (2012) Macrophage-derived angiopoietin-like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol., 32, 1400–1409.
22) Morinaga, J., Kadomatsu, T., Miyata, K., Endo, M., Terada, K., Tian, Z., Sugizaki, T., Tanigawa, H., Zhao, J., Zhu, S., et al. (2016) Angiopoietin-like protein 2 increases renal fibrosis by accelerating transforming growth factor-beta signaling in chronic kidney disease. Kidney Int., 89, 327–341.
23) Denk, D. & Greten, F.R. (2022) Inflammation: The incubator of the tumor microenvironment. Trends Cancer, 8, 901–914.
24) Greten, F.R. & Grivennikov, S.I. (2019) Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity, 51, 27–41.
25) Aoi, J., Endo, M., Kadomatsu, T., Miyata, K., Nakano, M., Horiguchi, H., Ogata, A., Odagiri, H., Yano, M., Araki, K., et al. (2011) Angiopoietin-like protein 2 is an important facilitator of inflammatory carcinogenesis and metastasis. Cancer Res., 71, 7502–7512.
26) Yuspa, S.H., Dlugosz, A.A., Cheng, C.K., Denning, M.F., Tennenbaum, T., Glick, A.B., & Weinberg, W.C. (1994) Role of oncogenes and tumor suppressor genes in multistage carcinogenesis. J. Invest. Dermatol., 103(Suppl), 90S–95S.
27) Kay, J., Thadhani, E., Samson, L., & Engelward, B. (2019) Inflammation-induced DNA damage, mutations and cancer. DNA Repair (Amst.), 83, 102673.
28) Reuter, S., Gupta, S.C., Chaturvedi, M.M., & Aggarwal, B.B. (2010) Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med., 49, 1603–1616.
29) Aoi, J., Endo, M., Kadomatsu, T., Miyata, K., Ogata, A., Horiguchi, H., Odagiri, H., Masuda, T., Fukushima, S., Jinnin, M., et al. (2014) Angiopoietin-like protein 2 accelerates carcinogenesis by activating chronic inflammation and oxidative stress. Mol. Cancer Res., 12, 239–249.
30) Moon, R.T., Kohn, A.D., De Ferrari, G.V., & Kaykas, A. (2004) WNT and beta-catenin signalling: Diseases and therapies. Nat. Rev. Genet., 5, 691–701.
31) Takahashi, M., Fukuda, K., Sugimura, T., & Wakabayashi, K. (1998) Beta-catenin is frequently mutated and demonstrates altered cellular location in azoxymethane-induced rat colon tumors. Cancer Res., 58, 42–46.
32) Horiguchi, H., Kadomatsu, T., Yumoto, S., Masuda, T., Miyata, K., Yamamura, S., Sato, M., Morinaga, J., Ohtsuki, S., Baba, H., et al. (2022) Tumor cell-derived ANGPTL2 promotes beta-catenin-driven intestinal tumorigenesis. Oncogene, 41, 4028–4041.
33) Petrova, V., Annicchiarico-Petruzzelli, M., Melino, G., & Amelio, I. (2018) The hypoxic tumour microenvironment. Oncogenesis, 7, 10.
34) Rankin, E.B., Nam, J.M., & Giaccia, A.J. (2016) Hypoxia: Signaling the Metastatic Cascade. Trends Cancer, 2, 295–304.
35) Elfrink, A.K.E., Nieuwenhuizen, S., van den Tol, M.P., Burgmans, M.C., Prevoo, W., Coolsen, M.M.E., van den Boezem, P.B., van Delden, O.M., Hagendoorn, J., Patijn, G.A., et al.; Dutch Hepato Biliary Audit Group; Collaborators. (2021) Hospital variation in combined liver resection and thermal ablation for colorectal liver metastases and impact on short-term postoperative outcomes: A nationwide population-based study. HPB (Oxford), 23, 827–839.
36) Lyssiotis, C.A. & Kimmelman, A.C. (2017) Metabolic interactions in the tumor microenvironment. Trends Cell Biol., 27, 863–875.
37) Valastyan, S. & Weinberg, R.A. (2011) Tumor metastasis: Molecular insights and evolving paradigms. Cell, 147, 275–292.
38) Fares, J., Fares, M.Y., Khachfe, H.H., Salhab, H.A., & Fares, Y. (2020) Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther., 5, 28.
39) Endo, M., Nakano, M., Kadomatsu, T., Fukuhara, S., Kuroda, H., Mikami, S., Hato, T., Aoi, J., Horiguchi, H., Miyata, K., et al. (2012) Tumor cell-derived angiopoietin-like protein ANGPTL2 is a critical driver of metastasis. Cancer Res., 72, 1784–1794.
40) Endo, M., Yamamoto, Y., Nakano, M., Masuda, T., Odagiri, H., Horiguchi, H., Miyata, K., Kadomatsu, T., Motokawa, I., Okada, S., et al. (2014) Serum ANGPTL2 levels reflect clinical features of breast cancer patients: Implications for the pathogenesis of breast cancer metastasis. Int. J. Biol. Markers, 29, e239–e245.
41) Simon, T., Gagliano, T., & Giamas, G. (2017) Direct effects of anti-angiogenic therapies on tumor cells: VEGF signaling. Trends Mol. Med., 23, 282–292.
42) Crawford, Y., Kasman, I., Yu, L., Zhong, C., Wu, X., Modrusan, Z., Kaminker, J., & Ferrara, N. (2009) PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell, 15, 21–34.
43) Xu, J., Lamouille, S., & Derynck, R. (2009) TGF-beta-induced epithelial to mesenchymal transition. Cell Res., 19, 156–172.
44) Masuda, T., Endo, M., Yamamoto, Y., Odagiri, H., Kadomatsu, T., Nakamura, T., Tanoue, H., Ito, H., Yugami, M., Miyata, K., et al. (2015) ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci. Rep., 5, 9170.
45) Muller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M.E., McClanahan, T., Murphy, E., Yuan, W., Wagner, S.N., et al. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature, 410, 50–56.
46) Zlotnik, A., Burkhardt, A.M., & Homey, B. (2011) Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol., 11, 597–606.
47) Osumi, H., Horiguchi, H., Kadomatsu, T., Tashiro, K., Morinaga, J., Takahashi, T., Ikeda, K., Ito, T., Suzuki, M., Endo, M., et al. (2020) Tumor cell-derived angiopoietin-like protein 2 establishes a preference for glycolytic metabolism in lung cancer cells. Cancer Sci., 111, 1241–1253.
48) Horiguchi, H., Endo, M., Miyamoto, Y., Sakamoto, Y., Odagiri, H., Masuda, T., Kadomatsu, T., Tanoue, H., Motokawa, I., Terada, K., et al. (2014) Angiopoietin-like protein 2 renders colorectal cancer cells resistant to chemotherapy by activating spleen tyrosine kinase-phosphoinositide 3-kinase-dependent anti-apoptotic signaling. Cancer Sci., 105, 1550–1559.
49) Pardal, R., Clarke, M.F., & Morrison, S.J. (2003) Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer, 3, 895–902.
50) Visvader, J.E. & Lindeman, G.J. (2012) Cancer stem cells: Current status and evolving complexities. Cell Stem Cell, 10, 717–728.
51) Huang, D., Sun, G., Hao, X., He, X., Zheng, Z., Chen, C., Yu, Z., Xie, L., Ma, S., Liu, L., et al. (2021) ANGPTL2-containing small extracellular vesicles from vascular endothelial cells accelerate leukemia progression. J. Clin. Invest., 131, 131.
52) Branco, M.R., Ficz, G., & Reik, W. (2011) Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet., 13, 7–13.
53) Itoh, H., Kadomatsu, T., Tanoue, H., Yugami, M., Miyata, K., Endo, M., Morinaga, J., Kobayashi, E., Miyamoto, T., Kurahashi, R., et al. (2018) TET2-dependent IL-6 induction mediated by the tumor microenvironment promotes tumor metastasis in osteosarcoma. Oncogene, 37, 2903–2920.
54) Crabtree, G.R. & Olson, E.N. (2002) NFAT signaling: Choreographing the social lives of cells. Cell, 109(Suppl), S67–S79.
55) Chinenov, Y. & Kerppola, T.K. (2001) Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene, 20, 2438–2452.
56) van Dam, H. & Castellazzi, M. (2001) Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene, 20, 2453–2464.
57) Kauffman, E.C., Ricketts, C.J., Rais-Bahrami, S., Yang, Y., Merino, M.J., Bottaro, D.P., Srinivasan, R., & Linehan, W.M. (2014) Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol., 11, 465–475.
58) Raben, N. & Puertollano, R. (2016) TFEB and TFE3: Linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol., 32, 255–278.
59) Horiguchi, H., Kadomatsu, T., Kurahashi, R., Hara, C., Miyata, K., Baba, M., Osumi, H., Terada, K., Araki, K., Takai, T., et al. (2019) Dual functions of angiopoietin-like protein 2 signaling in tumor progression and anti-tumor immunity. Genes Dev., 33, 1641–1656.
60) Kadomatsu, T., Hara, C., Kurahashi, R., Horiguchi, H., Morinaga, J., Miyata, K., Kurano, S., Kanemaru, H., Fukushima, S., Araki, K., et al. (2023) ANGPTL2-mediated epigenetic repression of MHC-I in tumor cells accelerates tumor immune evasion. Mol. Oncol., 17, 2637–2658.
61) Kurahashi, R., Kadomatsu, T., Baba, M., Hara, C., Itoh, H., Miyata, K., Endo, M., Morinaga, J., Terada, K., Araki, K., et al. (2019) MicroRNA-204-5p: A novel candidate urinary biomarker of Xp11.2 translocation renal cell carcinoma. Cancer Sci., 110, 1897–1908.
62) Wirtz, S., Popp, V., Kindermann, M., Gerlach, K., Weigmann, B., Fichtner-Feigl, S., & Neurath, M.F. (2017) Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc., 12, 1295–1309.
63) Horiguchi, H., Kadomatsu, T., Miyata, K., Terada, K., Sato, M., Torigoe, D., Morinaga, J., Moroishi, T., & Oike, Y. (2021) Stroma-derived ANGPTL2 establishes an anti-tumor microenvironment during intestinal tumorigenesis. Oncogene, 40, 55–67.
64) Tay, R.E., Richardson, E.K., & Toh, H.C. (2021) Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther., 28, 5–17.
65) Borst, J., Ahrends, T., Babala, N., Melief, C.J.M., & Kastenmuller, W. (2018) CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol., 18, 635–647.
66) Farhood, B., Najafi, M., & Mortezaee, K. (2019) CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol., 234, 8509–8521.
67) Raskov, H., Orhan, A., Christensen, J.P., & Gogenur, I. (2021) Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer, 124, 359–367.
68) Dunn, G.P., Koebel, C.M., & Schreiber, R.D. (2006) Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol., 6, 836–848.
69) Garrido, F. & Aptsiauri, N. (2019) Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology, 158, 255–266.
70) Jongsma, M.L.M., Guarda, G., & Spaapen, R.M. (2019) The regulatory network behind MHC class I expression. Mol. Immunol., 113, 16–21.
71) Kobayashi, K.S. & van den Elsen, P.J. (2012) NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol., 12, 813–820.
72) Burr, M.L., Sparbier, C.E., Chan, K.L., Chan, Y.C., Kersbergen, A., Lam, E.Y.N., Azidis-Yates, E., Vassiliadis, D., Bell, C.C., Gilan, O., et al. (2019) An evolutionarily conserved function of polycomb silences the MHC Class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell, 36, 385–401 e388.
73) Laugesen, A., Hojfeldt, J.W., & Helin, K. (2019) Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol. Cell, 74, 8–18.
74) van Mierlo, G., Veenstra, G.J.C., Vermeulen, M., & Marks, H. (2019) The Complexity of PRC2 Subcomplexes. Trends Cell Biol., 29, 660–671.
75) Martins, F., Sofiya, L., Sykiotis, G.P., Lamine, F., Maillard, M., Fraga, M., Shabafrouz, K., Ribi, C., Cairoli, A., Guex-Crosier, Y., et al. (2019) Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol., 16, 563–580.
76) Mahmood, S.S., Fradley, M.G., Cohen, J.V., Nohria, A., Reynolds, K.L., Heinzerling, L.M., Sullivan, R.J., Damrongwatanasuk, R., Chen, C.L., Gupta, D., et al. (2018) Myocarditis in patients treated with immune checkpoint inhibitors. J. Am. Coll. Cardiol., 71, 1755–1764.
77) Horiguchi, H., Kadomatsu, T., Yamashita, T., Yumoto, S., Terada, K., Sato, M., Morinaga, J., Miyata, K., & Oike, Y. (2023) ANGPTL2 promotes immune checkpoint inhibitor-related murine autoimmune myocarditis. Commun. Biol., 6, 965.
78) Kikuchi, R., Tsuda, H., Kozaki, K., Kanai, Y., Kasamatsu, T., Sengoku, K., Hirohashi, S., Inazawa, J., & Imoto, I. (2008) Frequent inactivation of a putative tumor suppressor, angiopoietin-like protein 2, in ovarian cancer. Cancer Res., 68, 5067–5075.
79) Peixoto, A., Relvas-Santos, M., Azevedo, R., Santos, L.L., & Ferreira, J.A. (2019) Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front. Oncol., 9, 380.
著者紹介Author Profile
 門松 毅(かどまつ つよし)
門松 毅(かどまつ つよし)熊本大学大学院生命科学研究部分子遺伝学講座 講師.博士(医学).
略歴2003年熊本大学理学部生物科学科卒業.09年同大学院医学教育部博士課程修了.10年熊本大学大学院医学薬学研究部助教.20年より現職.
研究テーマと抱負ANGPTL2シグナルやミトコンドリア恒常性の観点から,加齢および加齢関連疾患発症・進展の分子機構解明を目指し研究を行っている.
ウェブサイトhttp://www.kumamoto-u-molgene.jp
趣味ランニング.
 尾池 雄一(おいけ ゆういち)
尾池 雄一(おいけ ゆういち)熊本大学大学院生命科学研究部分子遺伝学講座 教授.博士(医学).
略歴1990年自治医科大学卒業.99年熊本大学大学院医学研究科博士課程(内科学専攻)修了.99年熊本大学医学部助教.2002年慶應義塾大学医学部専任講師.07年より現職.23年熊本大学医学部長(兼任),熊本大学大学院生命科学部長(兼任).
研究テーマと抱負循環器専門医,総合内科専門医,肥満症専門医として実臨床から生じる老化・加齢関連疾患に対する疑問を基礎医学研究により解明し,健康寿命延伸の具現化を目指している.
ウェブサイトhttp://www.kumamoto-u-molgene.jp
趣味健康ゴルフ.