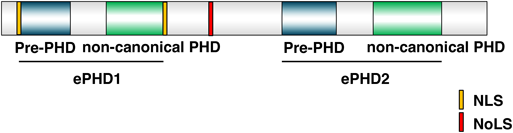
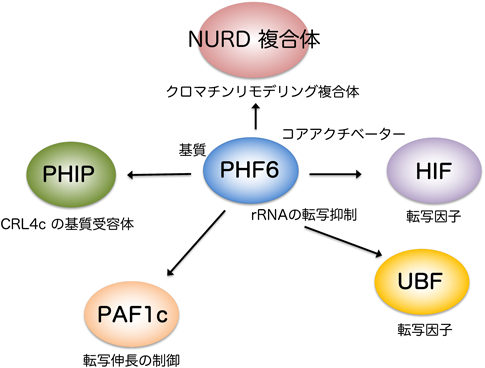
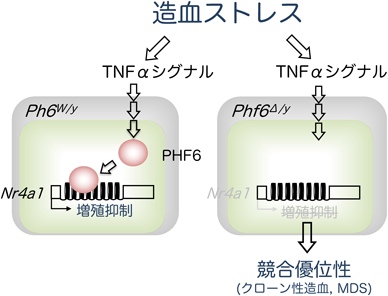
クロマチンタンパク質PHF6による造血幹細胞の制御Regulation of hematopoietic stem cell by chromatin protein, PHF6
島根大学医学部生化学講座Department of Biochemistry, Faculty of Medicine, Shimane University ◇ 〒693–0021 島根県出雲市塩冶町89-1 ◇ 89–1 Enya, Izumo, Shimane 693–0021, Japan
発行日:2024年6月25日Published: June 25, 2024