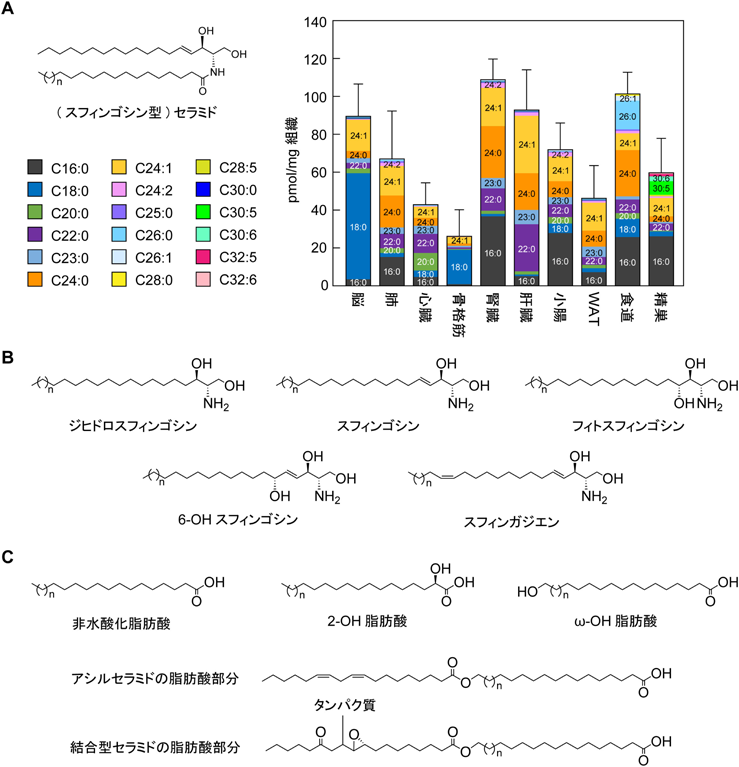
一般的な哺乳類組織におけるセラミドの組成は比較的単純である.長鎖塩基部分の大部分はC-4位とC-5位の間にトランス二重結合を持つスフィンゴシンである.脂肪酸部分の鎖長の多くはC16からC24であり,7大主要分子種はC16:0, C18:0, C20:0, C22:0, C23:0, C24:0, C24:1脂肪酸である2, 3)(図1A).これらのうち,C20:0, C22:0, C23:0, C24:0, C24:1脂肪酸はグリセロリン脂質にはほとんど含まれないことからスフィンゴ脂質に特徴的な脂肪酸といえる.セラミドの脂肪酸組成は組織ごとに大きく異なっている(図1A).たとえば脳や骨格筋にはC18:0脂肪酸含有セラミド(C18:0セラミド)が多いのに対して,肝臓にはC24:0セラミドやC24:1セラミドが多い.哺乳類には六つのセラミド合成酵素アイソザイム(CERS1からCERS6)が存在し,それぞれが基質であるアシルCoAの炭素鎖長に特異性を示す4).このセラミド合成酵素の組織ごとのアイソザイムの発現量の違いが,各組織に特徴的なセラミド組成を生み出す一因である.それぞれの鎖長の脂肪酸を含有するセラミド/複合スフィンゴ脂質の分子レベルでの役割については不明な点が多いが,組織レベルでの重要性は各セラミド合成酵素の遺伝子ノックアウト(knockout:KO)マウスの表現型あるいはヒトの遺伝性疾患の症状などから明らかとなってきている(後述).
セラミドに極性基としてホスホコリンあるいは糖/糖鎖が付加されると,それぞれスフィンゴミエリンあるいはスフィンゴ糖脂質と呼ばれる複合スフィンゴ脂質になる.スフィンゴ糖脂質中の糖/糖鎖にはグルコース,ガラクトース,シアル酸,N-アセチルグルコサミン,N-アセチルガラクトサミン,フコースなどがあり,これらの糖が一つから多いもので10以上含まれる1, 5).複合スフィンゴ脂質は形質膜の外層に多く存在し,特にスフィンゴミエリンはコレステロールとともに会合して局所的に流動性が低い領域(脂質ラフト)を形成する.複合スフィンゴ脂質に含まれるセラミド骨格部分を,より流動性が高い領域を形成するグリセロリン脂質のジアシルグリセロール骨格部分と比較すると,セラミドが水素結合や疎水性相互作用による脂質–脂質相互作用しやすい構造を持っていることがわかる.まず,複合スフィンゴ脂質中のセラミド部分は水素結合のドナーおよびアクセプターを持つのに対して,グリセロリン脂質中のジアシルグリセロール部分は水素結合のアクセプターしか持たない.二番目にセラミドの疎水鎖は長鎖塩基部分がトランス二重結合,脂肪酸部分の多くが飽和であることから折れ曲がりが少ない一方,ジアシルグリセロールにはシス二重結合を持つオレイン酸,リノール酸,α-リノレン酸,アラキドン酸などが含まれるため,折れ曲がった構造を持つ.この折れ曲がり構造は脂質–脂質相互作用を妨げる.三番目に,セラミドの脂肪酸部分にはC22やC24などの長い鎖長を持つ脂肪酸が多いのに対して,ジアシルグリセロールを構成する脂肪酸の炭素鎖長は主にC16からC20である6).セラミド中のこの長い脂肪酸鎖長は疎水性相互作用による脂質–脂質間相互作用を増強するだけではなく,そのω末端が形質膜の内層にまで達し,内層側での特異的な脂質やタンパク質の集積を引き起こす7).
ヒトの長鎖塩基にはスフィンゴシン以外にも,二重結合を持たないジヒドロスフィンゴシン,C-4位に水酸基を持つフィトスフィンゴシン,C-4位とC-5位の間のトランス二重結合(Z)とC-6位に水酸基を持つ6-水酸化(6-OH)スフィンゴシン,C-4位とC-5位の間のトランス二重結合とC-14位とC-15位の間にシス二重結合(E)を持つスフィンガジエンが存在する1, 4)(図1B).これらのうち,ジヒドロスフィンゴシンとスフィンガジエンを含有するセラミド(それぞれジヒドロセラミド,スフィンガジエン型セラミド)は量こそ少ないものの,広範な組織に存在する.ただし,ジヒドロセラミドは毛髪,スフィンガジエン型セラミドは脳や腎臓には多く存在する8, 9).フィトスフィンゴシン含有セラミド(別名フィトセラミド)は表皮,小腸,胃,腎臓などの上皮系の組織に存在する10–13).6-OHスフィンゴシン型セラミドは表皮や口腔などの角化扁平重層上皮にのみ存在する11, 12, 14).ただし,マウスには存在しない11).
ヒトのセラミドの脂肪酸部分としては一般的な脂肪酸である非水酸化脂肪酸に加えて,2-OH(α-OH)脂肪酸,ω-OH脂肪酸が存在する1, 4)(図1C).マウスにはさらに3-OH(β-OH)脂肪酸も存在する11).特定の組織のセラミドには特徴的な脂肪酸鎖長/不飽和度を持つ脂肪酸が存在する.たとえば,表皮と食道のセラミドにはC26:0の非水酸化脂肪酸が存在する11, 12, 14).また,精巣(精子)にはC30:5やC30:6などの多価不飽和の非水酸化脂肪酸が存在し,精子形成に重要な役割を果たしている2, 15)(図1A).C24:2非水酸化脂肪酸を含むセラミドは比較的広い組織にみられるが,特に脾臓に多い2).2-OH脂肪酸を含むセラミド/スフィンゴ脂質は脳のミエリン,胃,顎下腺に多い16, 17).ω-OH脂肪酸を含有するセラミド(ω-OHセラミド)は表皮と消化管上部(口腔,食道など)の扁平重層上皮に存在する11, 12, 14).ただし,ω-OHセラミドのほとんどはそのままの状態ではなく,ω位にリノール酸が結合した状態(アシルセラミド),あるいはそのリノール酸部分がエポキシエノン化修飾とタンパク質結合を受けた状態(結合型セラミド)で存在する1, 4)(図1C).ω-OHセラミド,アシルセラミド,結合型セラミド中のω-OH脂肪酸の鎖長はきわめて長く,C30からC36である11, 12).
多くの組織ではセラミドのほとんどは複合スフィンゴ脂質の合成あるいは分解の途中で産生される一過的な代謝中間体として存在するため,量はそれほど多くない(スフィンゴミエリン量の10分の1程度).一方,表皮ではセラミドは透過性バリアを形成するための脂質ラメラの主要な構成成分であるため,多様かつ多量(通常の組織の数十倍)に存在する.特にヒトの角質層のセラミドは多様性に富み,23クラス(異なる種類の長鎖塩基と脂肪酸の組合わせ)の1500を超える分子種(鎖長の違い)が存在する12).哺乳類の長鎖塩基の炭素鎖長はほとんどがC18であるが,脳,食道,胃などの一部の組織ではC20のものもみられる13, 18).また,例外的に表皮の長鎖塩基の炭素鎖長はC16からC26と幅広い12).表皮におけるセラミドの多様性,皮膚疾患とセラミド組成の関係,アシルセラミドと結合型セラミドの皮膚バリア形成における役割と産生の分子機構については大野らの稿を参照されたい.
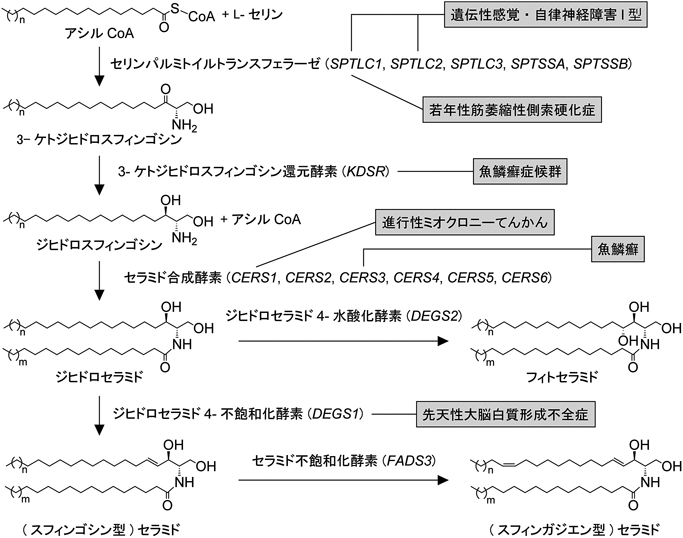
セラミド/スフィンゴ脂質の産生経路における最初の反応はL-セリンとアシルCoAの縮合による3-ケトジヒドロスフィンゴシン(3-ketodihydrosphingosine:KDS)の産生である19)(図2).この反応において使用されるアシルCoAのほとんどがC16:0のパルミトイルCoA(C16:0-CoA)であるため,この反応を触媒する酵素はセリンパルミトイルトランスフェラーゼ(serine palmitoyltransferase:SPT)と呼ばれる.C16:0-CoAがこの反応の基質として用いられる場合,L-セリンからC2単位が供給されるため,産生されるKDSの炭素鎖長はC18である.SPTは大サブユニットであるSPTLC1, SPTLC2, SPTLC3と小サブユニットSPTSSA, SPTSSBからなるヘテロオリゴマーであり,組合わせにはSPTLC1/2/SSA, SPTLC1/2/SSB, SPTLC1/3/SSA, SPTLC1/3/SSBの4通りがある20).このうち,SPTLC2とSPTLC3が補酵素であるピリドキサールリン酸と結合するリシン残基を有し,触媒活性を持つ19, 21).いずれの複合体もC18のKDSを産生する活性を示すが,C16のKDS産生にはSPTLC1/3/SSAとSPTLC1/3/SSB, C20のKDS産生にはSPTLC1/2/SSBとSPTLC1/3/SSB, C20より長いKDS産生にはSPTLC1/3/SSBが関わる12, 20).多くの組織で主に働いているのはSPTLC1/2/SSA複合体である.SPTLC1あるいはSPTLC2の特定のミスセンス変異は顕性遺伝様式の遺伝性感覚・自律神経障害I型を引き起こす22, 23).これらの変異によってSPTの基質ポケットの構造が変化し,通常の基質であるL-セリン以外にもL-アラニンやL-グリシンがSPTの基質として用いられることで,C-1位に水酸基を持たないデオキシ型のKDSが産生される.このデオキシ型KDSからは神経毒性を示すデオキシ型のセラミドが産生される.別のSPTLC1の変異は若年性筋萎縮性側索硬化症を引き起こす24).SPTの活性はORMDLタンパク質によって抑制的に制御されているが25),この変異によってORMDLタンパク質(ORMDL1から3の三つのアイソザイムが哺乳類には存在)による抑制効果が減弱し,スフィンゴ脂質の産生量が増加する24).
KDSはKDS還元酵素KDSR(別名FVT1)によって還元され,ジヒドロスフィンゴシンへ変換される26)(図2).KDSRの変異は血小板減少を伴う魚鱗癬症候群を引き起こす27, 28).魚鱗癬は表皮の過角化,乾燥,落屑を伴う皮膚疾患である.
ジヒドロスフィンゴシンはアシルCoAと縮合し,ジヒドロセラミドになる(図2).この反応はセラミド合成酵素(CERS1からCERS6)が触媒する4).セラミド合成酵素はジヒドロスフィンゴシンだけでなく,分解過程で生じた他の長鎖塩基も基質とすることができる.セラミド合成酵素は長鎖塩基に対しては明確な基質特異性を示さないが9),もう一つの基質であるアシルCoAに対しては高い基質特異性を示す.たとえば,CERS1はC18:0-CoAに対して活性が高い29).CERS1は脳(神経細胞)や骨格筋で発現が高く,これらの組織にC18:0セラミドが多いことと相関する2, 3, 30).Cers1 KOマウスあるいは自然誘発変異マウスでは小脳の異常(葉状構造の形成異常,進行性の縮小,プルキンエ細胞の変性)と協調運動の低下などがみられる31, 32).ヒトにおいてCERS1変異は進行性ミオクロニーてんかんを引き起こす33).CERS2はC22からC24までのアシルCoAに対して活性が高い30, 34).CERS2は広範な組織に発現しているが,特にC22/C24セラミドが多い肝臓,腎臓,脳のミエリンで高発現している30, 35).Cers2 KOマウスは肝障害,高頻度の肝がん,ミエリン形成異常,小脳変性といった表現型を示す36, 37).CERS3はC18以上の幅広いアシルCoAに活性を示すが,生理的には他のCERSが活性を示さないC26以上のアシルCoAに対する活性が重要である34, 38, 39).この活性によってCERS3は表皮におけるアシルセラミドと結合型セラミド,精子における多価不飽和セラミドの産生に関わる15, 39).CERS3の遺伝子発現はそれらの組織に限局している38).Cers3の全身KOマウスは皮膚バリア異常で新生致死となり,生殖細胞特異的なコンディショナルKOマウスは雄性不妊を示す15, 39).また,ヒトにおいてCERS3変異は魚鱗癬を引き起こす40).CERS4はC18:0-CoA, C20:0-CoA, C22:0-CoAに活性を示す34, 41).CERS4は比較的多くの組織で発現しているが30, 41),特に皮脂腺での発現が高く,Cers4 KOマウスは進行性の脱毛を示す42).CERS5とCERS6はともにC16:0-CoAに対して活性を示し,広範な組織で発現している30, 34, 41, 43).これらは重複した機能を持つため,それぞれのKOマウスは通常条件下では明確な表現型を示さないが,Cers5 KOマウスは高脂肪食による肥満になりづらく44),Cers6 KOマウスは自己免疫性脳脊髄炎モデルにおいて炎症の増悪を示す45).
ジヒドロセラミドのジヒドロスフィンゴシン部分のC-4位とC-5位の間にトランス二重結合が導入されるとスフィンゴシン型のセラミド,C-4位に水酸基が導入されるとフィトセラミドになる(図2).ジヒドロセラミド4-不飽和化酵素DEGS1はスフィンゴシン型セラミド産生において中心的な役割を果たす46, 47).一方,ジヒドロセラミド4-水酸化酵素DEGS2はフィトセラミド産生を触媒する46).DEGS2はジヒドロセラミド4-不飽和化酵素としても働く二機能酵素である.Degs1 KOマウスではスフィンゴシン型セラミドの量が野生型マウスの約10分の1に減少する47).この残存した分はDEGS2が産生していると考えられる.DEGS2はC16やC18などの短い脂肪酸を持つジヒドロセラミドに対しては4-不飽和化活性が高く,C22以上の長い脂肪酸を持つジヒドロセラミドに対しては4-水酸化活性が高い13).Degs1 KOマウスは低い出生率,小さい体躯,魚鱗癬様の皮膚とまばらな毛,骨塩と密度の低下,肝臓の機能低下を示し,8から10週で死亡する47).ヒトにおいてDEGS1変異は先天性大脳白質形成不全症を引き起こす48).Degs2 KOマウスではフィトセラミド量が野生型マウスの約6分の1に減少するが,消失はしない13).そのため,DEGS2以外にもジヒドロセラミド4-水酸化活性を持つ酵素が存在すると考えられる.フィトセラミドは上皮系の組織に存在し,C-4位水酸基を介した水素結合による脂質–脂質相互作用によって透過性バリアを増強する役割があると想定されるが,Degs2 KOマウスの皮膚や消化器における透過性バリアは正常である13).この原因は,ヒトと異なってフィトセラミドの量がもともとマウスには少ないこと,他の4-水酸化酵素が存在していること,マウスには他の水酸化セラミド(脂肪酸のβ位あるいはω位に水酸基を持ったセラミド)が多く存在して機能を代替しているなどの可能性が考えられる11, 13).
スフィンゴシン型セラミドのスフィンゴシン部分のC-14とC-15位の間にシス二重結合が導入されるとスフィンガジエン型セラミドになる(図2).この反応はセラミド14-不飽和化酵素FADS3が触媒する9, 49).上述のDEGS1とDEGS2はそれぞれ別名がFADS7, FADS8であり,FADS3とともにFADS(fatty acid desaturase)ファミリーに属す.FADSファミリーの中でFADS1, FADS2, FADS4, FADS5がアシルCoAを基質とするのに対し,FADS3, FADS7/DEGS1, FADS8/DEGS2はセラミド(FADS3,スフィンゴシン型セラミド;DEGS1とDEGS2,ジヒドロセラミド)を基質とする46, 50).
6-OHスフィンゴシン型セラミドの産生経路は不明であるが,スフィンゴシン型セラミドのC-6位が未知の水酸化酵素によって水酸化されて産生されると考えられる.長鎖塩基の中でde novo合成経路によって産生されるのはジヒドロスフィンゴシンのみである.他の長鎖塩基はセラミドのセラミダーゼによる分解によってのみ産生する.分解によって生じた各種長鎖塩基はセラミド合成酵素によってセラミド合成に再利用されるか,あるいは下記に記載の分解経路によってアシルCoAへ変換されて脂肪酸β酸化またはグリセロ脂質などの脂質合成に利用される.
形質膜の構成脂質である複合スフィンゴ脂質は,常に一部がエンドサイトーシスによってリソソームに運ばれ,分解されることで量の恒常性が保たれている.複合スフィンゴ脂質はリソソームの加水分解酵素によってセラミドに変換された後,セラミダーゼ(酸性セラミダーゼASAH1)によって長鎖塩基と脂肪酸になる.セラミダーゼは至適pHによって酸性(ASAH1),中性(ASAH2),アルカリ性セラミダーゼ(ACER1, ACER2, ACER3)に分類される51).これらのうち生理的に最も重要なのは酸性セラミダーゼASAH1であり,Asah1 KOマウスは胚性致死となる52).ヒトのASAH1変異は脂肪性の肉芽腫形成,関節腫脹変形,皮下結節などを伴うファーバー脂肪肉芽腫症(ファーバー病)あるいは進行性のミオクロニーてんかんを伴う脊髄性筋萎縮症を引き起こす53, 54).中性セラミダーゼASAH2は形質膜に局在するか,あるいは切断を受けて細胞外へ分泌される55).食事中のスフィンゴ脂質は長鎖塩基と脂肪酸に分解されないと小腸に吸収されづらい.ASAH2は食事由来のセラミドの小腸での分解に関わる56).
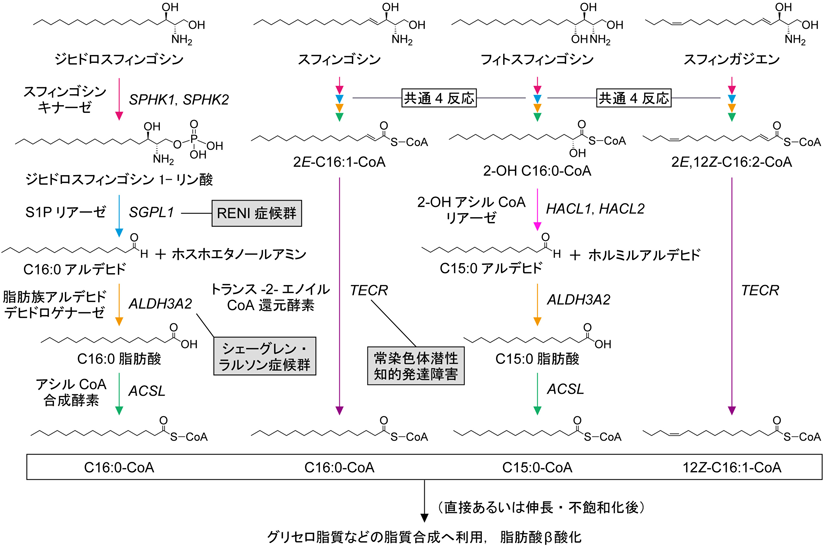
長鎖塩基のアシルCoAへの代謝過程ではいずれの長鎖塩基もC-1位のリン酸化(長鎖塩基1-リン酸の産生),C-2位とC-3位の間の開裂(長鎖アルデヒドの産生),酸化(長鎖脂肪酸の産生),CoA付加(アシルCoAの産生)という共通4反応を受ける4, 57)(図3).これまでに6-OHスフィンゴシン以外の長鎖塩基について詳細な代謝経路と反応が解明されているが,そのうち,筆者らはスフィンゴシン,フィトスフィンゴシン,スフィンガジエンの代謝経路の全体像の解明と,酸化以降の反応に関わる遺伝子の同定を行った58–62).特にフィトスフィンゴシンの過程においては脂肪酸α酸化と呼ばれるユニークな奇数鎖脂肪酸を生み出す脂肪酸の代謝経路があることを明らかにした17, 60, 62).
長鎖塩基は炭素数,水酸基の数(二つの場合d,三つの場合t),二重結合の数によって表記することができる.たとえば,哺乳類に最も多い炭素数C18の場合,ジヒドロスフィンゴシンはd18:0,スフィンゴシンはd18:1(二重結合の位置と種類を記載する場合は4E-d18:1),フィトスフィンゴシンはt18:0, 6-OHスフィンゴシンはt18:1(4E-t18:1),スフィンガジエンはd18:2(4E,14Z-d18:2)である.下記では,これらのC18を持つ長鎖塩基の代謝を例にして説明する.
d18:0ジヒドロスフィンゴシンは共通の4反応によって炭素数を2減じたC16:0-CoAに変換される59).一方,C-4位とC-5位の間にトランス二重結合を持つ4E-d18:1スフィンゴシンは2E-C16:1-CoA(トランス-2-ヘキサデセノイルCoA)になった後,還元(飽和化)によってC16:0-CoAになる59, 61, 63)(図3).一段階目のリン酸化はスフィンゴシンキナーゼ(SPHK1, SPHK2)によって触媒され,ジヒドロスフィンゴシンとスフィンゴシンからそれぞれジヒドロスフィンゴシン1-リン酸とスフィンゴシン1-リン酸(sphingosine 1-phosphate:S1P)が産生される64, 65).このうち,S1Pは血漿やリンパ液などの細胞外液に含まれる有名な脂質メディエーターである66, 67).S1Pは細胞表面のS1P受容体(S1PR1からS1PR5)に結合することで細胞運動,接着結合の増強,細胞増殖,アクチンの再構成などさまざまな細胞応答を引き起こし,特に血管形成やTリンパ球のリンパ組織からの移出において重要な役割を果たす.SPHK1とSPHK2は重複した機能を持つため,それぞれの単独のKOマウスは明らかな表現型を示さないが,Sphk1 Sphk2二重KOマウスは脂質メディエーターS1Pの産生不全のため,胎生致死となる68).S1Pは赤血球や内皮細胞などの一部の細胞からトランスポーターを介して細胞外へ排出される69, 70).一方,それ以外での細胞ではS1Pは細胞外に放出されることなく,細胞内で長鎖塩基代謝経路によって代謝されるか,S1Pホスファターゼによる脱リン酸化によってスフィンゴシンに戻る4).進化の過程でS1P受容体が出現するのは脊索動物以降であるが,長鎖塩基代謝に関わる各酵素は真核生物すべてに備わっている.このことから,S1Pは進化の過程で後から脂質メディエーターとしての機能を獲得したと考えられる.
長鎖塩基代謝の2段階目の開裂反応によって,d18:0ジヒドロスフィンゴシン1-リン酸と4E-d18:1 S1Pは長鎖アルデヒド(それぞれC16:0アルデヒドと2E-C16:1アルデヒド)とホスホエタノールアミンになる(図3).この反応は不可逆反応であり,小胞体に局在するS1PリアーゼSGPL1が触媒する71, 72).Sgpl1 KOマウスは肺,心臓,尿路,骨における形態異常,肝臓における脂質代謝異常,末梢におけるリンパ球減少,骨髄腫細胞の過形成などの表現型を示し,生後50日以内に死亡する73, 74).ヒトにおけるSGPL1遺伝子変異はステロイド抵抗性ネフローゼ症候群(RENI症候群;renal, endocrine, neurologic, and immune syndrome)を引き起こす75, 76).RENI症候群では進行性の腎機能障害,魚鱗癬,副腎機能不全,免疫不全および神経障害などの多様な症状がみられる.SGPL1は長鎖塩基代謝経路の律速段階の開裂反応を触媒する唯一の酵素であるため,その機能不全によって引き起こされる表現型/症状の大部分はスフィンゴ脂質/セラミドの恒常性の破綻に起因していると考えられる.ただし,末梢におけるリンパ球減少などのSgpl1 KOマウスの一部の表現型は,脂質メディエーターとしてのS1Pの機能に関連している可能性が高い.Tリンパ球のリンパ組織からの移出には,リンパ組織と血液間のS1Pの濃度勾配が重要であり,Sgpl1のKOによるリンパ組織内のS1P濃度上昇はそのリンパ組織–血液間の濃度勾配を減少させるからである.SGPL1によって産生されるもう一つの生成物であるホスホエタノールアミンは主にグリセロリン脂質であるホスファチジルエタノールアミンの産生に用いられる77).
長鎖塩基代謝の3段階目の酸化反応は脂肪族アルデヒドデヒドロゲナーゼ(fatty aldehyde dehydrogenase:FALDH)によって触媒される58)(図3).この反応によってd18:0ジヒドロスフィンゴシンと4E-d18:1スフィンゴシン由来の長鎖アルデヒドはそれぞれ,C16:0脂肪酸(パルミチン酸)と2E-C16:1脂肪酸(トランス-2-ヘキサデセン酸)に変換される.FALDHとしてヒトにはALDH3A1(サイトゾル),ALDH3A2(小胞体),ALDH3B1(形質膜)が存在し,マウスにはさらにALDH3B2(脂肪滴)とALDH3B3(形質膜)が存在する78, 79).これらのうち,長鎖塩基代謝には小胞体に局在するALDH3A2が最も高い寄与を示す58).ヒトにおけるALDH3A2変異は魚鱗癬,精神遅滞,痙性対麻痺あるいは四肢麻痺を主症状とするシェーグレン・ラルソン症候群(Sjögren–Larsson syndrome:SLS)を引き起こす80).SLSでは他のFALDHの活性が残るため,長鎖塩基の代謝は完全には停止しない.したがって,SLSでみられる症状は長鎖塩基からアシルCoAへの代謝不全が原因というよりも,蓄積した長鎖アルデヒドの毒性が原因と考えられている.Aldh3a2 KOマウスはヒトには存在しないAldh3b2との機能重複性から明確なSLS様の表現型を示さないが,軽微な運動機能異常を示す63, 81).一方,Aldh3a2 Aldh3b2二重KOマウスは透過性バリア異常を伴う魚鱗癬様の皮膚表現型を示す82).
長鎖塩基代謝の4段階目の脂肪酸のCoA付加反応はアシルCoA合成酵素(Acyl-CoA synthetase:ACS)が触媒する(図3).この反応によってd18:0ジヒドロスフィンゴシンと4E-d18:1スフィンゴシン由来の長鎖脂肪酸はそれぞれC16:0-CoAと2E-C16:1-CoAになる.ヒトには26種類のACSが存在するが,その中でも長鎖脂肪酸に高い活性を示すACSLサブファミリーメンバーが長鎖塩基代謝に主に関わる59).
スフィンゴシン由来の2E-C16:1-CoAは還元されてC16:0-CoAになる(図3).この反応はトランス-2-エノイル還元酵素TECRが触媒する61).TECRは長鎖塩基代謝だけでなく,小胞体における脂肪酸伸長サイクルにも関わる83).このサイクルは縮合,還元,脱水,還元の4反応からなり,1サイクルごとにアシルCoAの炭素数を二つ増加させる84).TECRはこのサイクルの最後の還元反応を触媒する.Tecr KOマウスは胚性致死を示す85).ヒトにおいてTECRの機能低下型のミスセンス変異(P182L)は非症候性の常染色体潜性知的発達障害を引き起こす86).この疾患の症状およびKOマウスの胚性致死性は長鎖塩基代謝異常が原因ではなく,主に脂肪酸伸長サイクルの異常に起因すると考えられる.
t18:0フィトスフィンゴシンは上記の共通4反応によって2-OH C16:0-CoAに変換される(図3).2-OHアシルCoAを脂肪酸β酸化あるいはスフィンゴ脂質以外の脂質合成に利用するためには2-水酸基の除去が必要である.哺乳類では3反応からなる脂肪酸α酸化によって2-水酸基の除去が行われる.この脂肪酸α酸化によって2-OH C16:0-CoAは1炭素短くなった非水酸化のC15:0-CoAに変換される87, 88).脂肪酸α酸化の最初の反応は2-OH C16:0-CoAの開裂反応であり,C15:0アルデヒドとホルミルアルデヒドが生じる89).この反応は2-OHアシルCoAリアーゼが触媒する.哺乳類にはHACL1(ペルオキシソーム)とHACL2(小胞体)の二つの2-OHアシルCoAリアーゼアイソザイムが存在し,フィトスフィンゴシン代謝にはこれらが重複して働く62, 90).2段階目の反応はFALDHによるC15:0アルデヒドからC15:0脂肪酸への酸化であり,FALDHの中でもALDH3A2の寄与が大きい62).3段階目の反応はACSによるC15:0脂肪酸からC15:0-CoAへの変換であり,主にACSLサブファミリーが働くと考えられる.脂肪酸α酸化はフィトスフィンゴシンの代謝産物である2-OH C16:0-CoAだけでなく,脂肪酸2-水酸化酵素(FA2H)によって産生された2-OH脂肪酸の分解においても働く17).上述のように哺乳類には2-OH脂肪酸を持つセラミド/スフィンゴ脂質が存在し,特に脳のミエリン,胃,顎下腺に多い17).セラミドの7大主要分子種のうちの一つであるC23:0セラミド中のC23:0脂肪酸は,2-OH C24:0脂肪酸からこの脂肪酸α酸化によって主に産生されたものである17).このような長い炭素鎖長の2-OH脂肪酸の代謝にはHACLのうち,HACL2が主要に働く.
4E,14Z-d18:2スフィンガジエンはスフィンゴシンと同じ5反応(共通4反応+還元/飽和化反応)によって,12Z-C16:1-CoAに変換される91)(図3).この代謝過程ではもともとC-4位にあったトランス二重結合は飽和化されるが,C-14位にあったシス二重結合は飽和化されない.そのため,スフィンガジエンは代謝によってω4系列という変わった一価不飽和脂肪酸へ変換される.
長鎖塩基の代謝経路によって産生されたアシルCoAはそのまま,あるいは脂肪酸伸長サイクルによる脂肪酸伸長または∆9不飽和化酵素による不飽和化を受けた後に多くはグリセロ脂質へ代謝される63, 91).スフィンゴ脂質の恒常性維持という点で長鎖塩基代謝経路は重要である74).一方,グリセロ脂質へのアシルCoAの供給という点では長鎖塩基代謝経路はマイナーな経路のため,長鎖塩基代謝経路が停止してもグリセロ脂質の量や組成には変化を与えない.