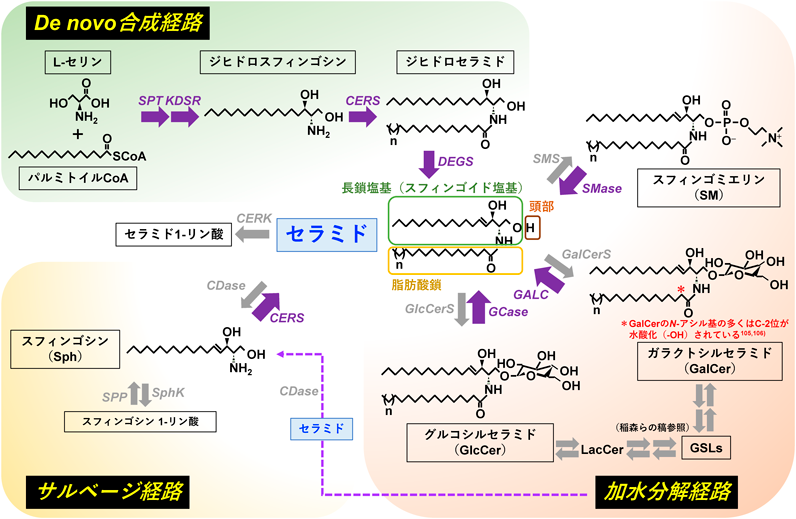
セラミドは「スフィンゴイド塩基(sphingoid base)」と呼ばれる長鎖塩基を骨格として持つ「スフィンゴ脂質(sphingolipids)」の一つであり,スフィンゴイド塩基のアミノ基にアシル基がアミド結合した構造を有する(図1)1).このセラミドにさまざまな親水性の頭部が結合したものを複合スフィンゴ脂質と呼び,糖/糖鎖やホスホコリン,リン酸などが結合することで,それぞれスフィンゴ糖脂質(glycosphingolipids:GSLs),スフィンゴミエリン(sphingomyelin:SM),セラミド1-リン酸(ceramide 1-phosphate:C1P)となる.また,セラミドのアミノ基が切断されて生じたスフィンゴイド塩基の一種であるスフィンゴシン(sphingosine:Sph)の頭部がリン酸化されるとスフィンゴシン1-リン酸(sphingosine 1-phosphate:Sph1P)が産生される.これらのスフィンゴ脂質は細胞膜構成脂質,または生理活性脂質として働く.セラミドにはスフィンゴイド塩基や脂肪酸部分の種類が異なるクラスが含まれ,それぞれのクラスには炭素鎖長の異なる多くの分子種が含まれる(大野らの稿を参照).脂肪酸部分の多様性は基質となるアシルCoAに異なる特異性を持つ6種類のセラミド合成酵素(ceramide synthase 1–6:CERS1–6)によって生み出される4–6)(木原の稿を参照).アシルセラミドは脂肪酸部分のω位にリノール酸がエステル結合した特殊なセラミドであり,皮膚の透過性バリア形成において重要な働きを示す(大野らの稿を参照).
セラミドの生理活性脂質としての機能は,1987年にBellとHannunらがリゾスフィンゴ脂質やスフィンゴイド塩基にプロテインキナーゼC(protein kinase C:PKC)阻害効果があることを発見した7, 8)ことがきっかけとなり,1989年に岡崎らによって,ヒト白血病細胞HL-60へのビタミンD処理によってSM分解からセラミドが産生し,そのセラミドが単球分化誘導を促進することを報告したことから始まる9).ほぼ同時期に,ホルボールエステル(PMA)処理による分化誘導でセラミドからのSM合成が亢進し,細菌由来のスフィンゴミエリナーゼ(sphingomyelinase:SMase)処理で細胞内のセラミドを増加させるとその分化が抑制されることから,セラミドやSMの生理活性脂質としての可能性が見いだされた10, 11).そして1993年にObeidらによって,細胞への短鎖セラミド(C2:0-セラミド,N-acetyl sphingosine:膜透過性の人工的なセラミド)処理によって「核の断片化」,「DNAラダー形成」など細胞死(アポトーシス)の兆候がみられたことが報告され12),セラミドには細胞死を誘導する「生理活性脂質」としての機能も見いだされた.こうしてセラミドによるシグナル伝達制御機構という概念が広まり,現在では,アポトーシス以外の細胞死(小木曽らの稿を参照),分化,老化,細胞骨格制御,インスリンシグナル抑制など,セラミドが関与する細胞内シグナル伝達制御機構の報告は増え続けている3, 13).
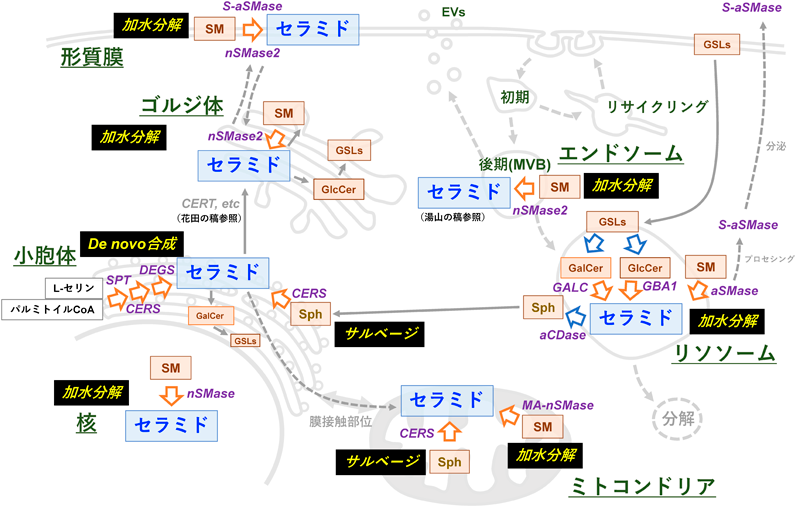
スフィンゴ脂質が命名された当初はSphを起点としてアシル化されセラミドが合成されると考えられていたが,その後,セラミドの産生経路には3種類あることが明らかとなってきた1).その3種類の産生経路は,①アミノ酸であるL-セリンと脂肪酸由来のパルミトイルCoAを起点とした“de novo合成経路”,②SM, GSLs, Sph1Pなどの分解からSphが産生され,そのSphがCERSの反応を受けてセラミドに再利用される“サルベージ経路”,③SMやGSLsの分解によって生じる“加水分解経路”である(図1).③において,SMはセラミドとホスファチジルコリンからSM合成酵素(SM synthase:SMS)により合成された後,SMaseにより加水分解されてホスホコリンとセラミドとなる.GSLsの合成ではまずセラミドにグルコースまたはガラクロースが結合し,グルコシルセラミド(glucosylceramide:GlcCer)やガラクトシルセラミド(Galactosylceramide:GalCer)が合成される(稲森らの稿を参照).これらのGlcCerやGalCerはグルコシルセラミダーゼ(glucosylceramidase:GCase)やガラクトシルセラミダーゼ(galactosylceramidase:GALC)によってグルコースやガラクトースが加水分解されてセラミドとなる14, 15).加水分解経路で産生されたセラミドの一部はさらにSphに分解されてからサルベージ経路を介してセラミド産生に利用される16).ここでは,各細胞小器官における3経路を介したセラミド産生について記載する(図2).
1)小胞体(endoplasmic reticulum:ER)
セラミドのde novo合成は,小胞体膜上でL-セリンとパルミトイルCoAがセリンパルミトイル転移酵素(serine palmitoyltransferase:SPT)により縮合して3-ケトジヒドロスフィンゴシン(3-ketodihydrosphingosine)が生成することから始まり,多段階を踏んでセラミドが合成される(木原の稿を参照).3-ケトジヒドロスフィンゴシンは,3-ケトジヒドロスフィンゴシン還元酵素(3-ketodihydrosphingosine reductase:KDSR)によりスフィンゴイド塩基骨格となるジヒドロスフィンゴシン(dihydrosphingosine)に変換される17).その後,CERSによってジヒドロスフィンゴシンのアミノ基にさまざまな炭素鎖長を持つ脂肪酸がアミド結合し,ジヒドロセラミド(dihydroceramide)が生成される.上述のように,CERSには6種類のアイソザイム(CERS1–6)が存在し,それぞれが基質であるアシルCoAの脂肪酸鎖長に対して異なる特異性を持つため,セラミドに分子種多様性を与えている4–6).最後にジヒドロセラミドのC-4,5位がジヒドロセラミド不飽和化酵素(dihydroceramide desaturase:DEGS)により不飽和化されセラミドとなる18).また,小胞体はCERSがSphを経てセラミドを合成するサルベージ経路の一端も担っている.短鎖セラミド(C2:0-セラミドなど)の細胞への処理では,小胞体でSphを経たサルベージ経路での内在性セラミド合成も起こることが知られている16).
2)ゴルジ体
ゴルジ体は細胞内物質の「修飾」や「輸送」に関わる.小胞体でde novo合成やサルベージ経路により産生されたセラミドは,セラミド輸送タンパク質(ceramide transporter:CERT)などによってゴルジ体に輸送されて,SMやグルコシルセラミドなどのGSLsの合成に利用された後,各生体膜へ輸送される(花田の稿を参照).また,ゴルジ体ではセラミドキナーゼ(ceramide kinase:CERK)によってC1Pが産生されることも報告されている19).他方,ゴルジ体にはnSMase2(遺伝子名はSMPD3)が局在していることが報告されている20).nSMase2を過剰発現した細胞において,サブコンフルエントの状態ではnSMase2はゴルジ体と形質膜に局在し,コンフルエントになるとゴルジ体から形質膜へ移行する20).また,ゴルジ体のnSMase2はサイトカインである腫瘍壊死因子α(tumor necrosis factor-α:TNF-α)の刺激に応じて形質膜に移動する.他方,PMA刺激ではnSMase2が形質膜からゴルジ体へ戻ることも報告されているが21),その意義についてはよくわかっていない.
3)リソソーム
リソソームは細胞内外の分子の分解に関わる細胞小器官であり,セラミド代謝においてもSMのホスホコリン部分やGSLsの糖鎖部分が除去されてセラミドが産生される.リソソームには至適pHが酸性領域のSMase(acid sphingomyelinase:aSMase:遺伝子名はSMPD1)が存在し,SMを加水分解する22).また,aSMaseの一部はプロセシングを受けた分泌型aSMase(secreted aSMase:S-aSMase)として,オートクリンまたはパラクリン的に形質膜のSM加水分解も引き起こす23).GlcCerはリソソーム局在性GCaseであるGBA1によって,GalCerはGALCによってセラミドへ変換される14, 15).リソソーム性セラミドは生理活性脂質として働くこともあるが,通常はセラミドの脂肪酸部分が酸性セラミダーゼ(acid ceramidase:aCDase)によって分解され,Sphが産生された後に再度小胞体に輸送され,CERSによってセラミドに再合成される(サルベージ経路)16).
4)エンドソーム
エンドソームは細胞内に取り込まれる(エンドサイトーシス)物質や,分泌される(エキソサイトーシス)物質を選別する場として機能する細胞小器官である.形態的な特徴や機能的な特徴から初期エンドソーム,後期エンドソーム,リサイクリングエンドソームに分けられる.エンドサイトーシスで取り込まれた物質はまず細胞辺縁部位の初期エンドソームに輸送され,その後,再利用されるものはリサイクリングエンドソームへ,分解経路へ進む場合は後期エンドソームからリソソームへ運ばれる.マウスリンパ腫細胞WR19Lにおいて,正常状態ではトランスフェリン(transferrin:Tf)刺激を受けると,Tf受容体はリサイクリングエンドソームを介して形質膜へ戻る24).しかし,SMS欠損細胞では形質膜SMの量が低下し,セラミドの割合が増えることで,Tf/Tf受容体が後期エンドソームからリソソームへ運ばれて分解するため,Tfによる細胞増殖が抑制されることが報告されている24).また,後期エンドソームは多胞体(multivesicular body:MVBs)とも呼ばれ,細胞外小胞(extracellular vesicles:EVs)の一種であるエクソソームの放出などに関わっている.nSMase2によるMVBsでのセラミド産生がエクソソームの放出に重要であることも示されている(湯山の稿を参照)25).
5)ミトコンドリア
ミトコンドリアのセラミドは小胞体からミトコンドリアとの接触部位を介して輸送されており,定常状態のミトコンドリアセラミド量は厳密に制御されている26).さまざまな刺激に応じて,小胞体でのセラミド産生が変化することで,ミトコンドリア膜のセラミド量が上昇し,アポトーシスやマイトファジーを誘導することが報告されている(小木曽らの稿を参照)26).また,マウス細胞においては,ミトコンドリア関連中性スフィンゴミエリナーゼ(mitochondria-associated neutral sphingomyelinase:MA-nSMase)が存在しており,SM加水分解によりセラミドを産生するが,ヒトではMA-nSMaseのオルソログをコードする遺伝子(SMPD5)は偽遺伝子である27).
6)形質膜
細胞内外を隔てる生体膜は「形質膜(plasma membrane)」と呼ばれる脂質二重膜である.リン脂質やスフィンゴ脂質が主要な構成脂質であるが,この形質膜においてもSM加水分解を介したセラミド産生が知られている3).特に,nSMase2によって内層側にセラミドが産生される経路と,S-aSMaseが細胞外層でセラミドを産生する経路が知られている25, 28).
7)核
核内にもセラミド産生経路が存在することが報告されている29–33).細胞から単離した核においてセラミドとSMが検出され,nSMaseによるSM加水分解によってセラミド産生が起こる29, 34, 35)とされているが,酵素の詳細には議論の余地が残っている.Albiらはクロマチン画分にSMとRNAを検出していることから,核膜中のSMが濃縮された微小領域に特定のクロマチンとRNAが会合している可能性がある36–38).ただし,核膜でのセラミドやSMの役割や分布についてはいまだ不明な点が多い.
以上,さまざまな細胞小器官で3種類の経路からセラミドが産生されている.「セラミドがどこで産生されるか?」は後のセラミドの生理活性脂質としての細胞生理作用の分子メカニズムを理解する上で非常に重要である.
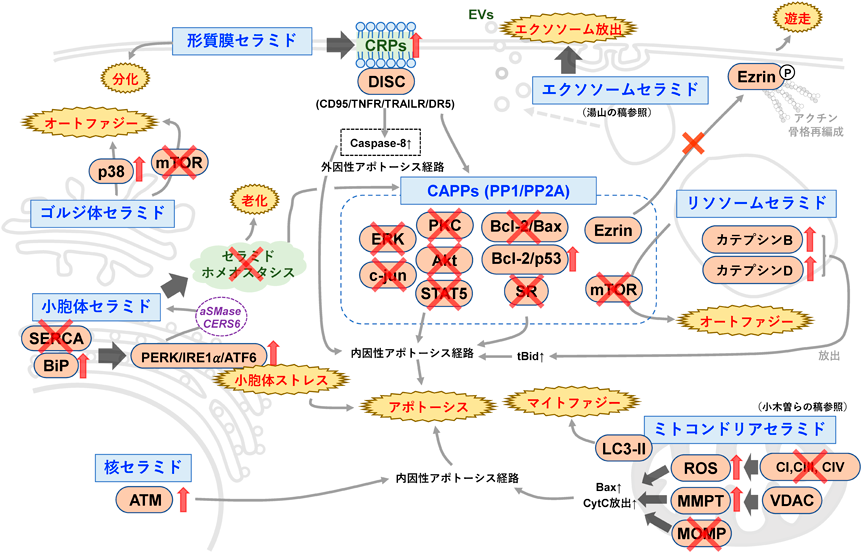
さまざまな細胞ストレスがセラミド上昇を伴った細胞死の引き金となることが次々と報告され,「セラミドの増加」=「細胞死」という認識は一般的になりつつある12, 39, 40).また,細胞内セラミド産生が増加し,セラミドが蓄積するとさまざまな細胞生理作用が引き起こされることは周知の事実である.ここでは「セラミドが産生される細胞小器官」に焦点を当ててセラミドの生理活性脂質としての機能とその分子メカニズムについて述べる(図3).生体内に天然に存在する長鎖セラミドは細胞に添加しても,膜透過性が悪く,細胞へはほとんど取り込まれない.そのため,セラミドの生理活性脂質機能発見の契機となったC2:0-セラミドのような膜透過性の短鎖セラミドは,細胞内セラミド作用を模倣するために広く利用されている(小木曽らの稿を参照).しかし,その性質や代謝は天然セラミドと異なる場合もあるため,その生理作用についての解釈には注意が必要である.
1)ミトコンドリア膜セラミド
先に述べたヒト白血病細胞HL-60へのC2:0-セラミド処理ではその後の研究から,活性酸素種(reactive oxygen species:ROS)の発生,ミトコンドリア機能不全,ミトコンドリア外膜透過性(mitochondrial outer membrane permeability:MOMP)の崩壊を経た内因性アポトーシスが誘導されることがわかってきた26).これらの短鎖セラミドを含むセラミド分子種によるミトコンドリアを介したアポトーシス誘導については別途稿があるためそちらを参照していただきたい(小木曽らの稿を参照).また,最近の報告では,ミトコンドリアのセラミドが電位依存性陰イオンチャネル2(voltage-dependent anion channel 2:VDAC2)と直接相互作用して,MOMPを引き起こすBaxの放出に関連することも示されている41).同様に,C1Pの産生に関わるCERKの阻害によって,肺がん細胞においてVDACを介したMMPT上昇とROS産生からシスプラチンの感受性の増加による細胞死が誘導されることが報告されているが,セラミドとの関連はわかっていない42).他にも,虚血再灌流刺激や放射線障害において誘導される内因性アポトーシス経路の活性化において,小胞体に局在するCERS1, CERS2, CERS6によるセラミド合成増加が,ミトコンドリアのセラミド蓄積に関連することが報告されている43, 44).他方,ミトコンドリアのC18:0-セラミドはマイトファジーにも関与することが報告されている(小木曽らの稿を参照)45, 46).
2)小胞体膜セラミド
小胞体ではde novo経路やサルベージ経路によってセラミドが合成される.しかしながら,このセラミド合成のバランスが崩れることでセラミドが蓄積し小胞体ストレスを介した細胞死が誘導される47).小胞体ストレス下では,細胞はunfolded protein response(UPR)を発動して正常状態を維持しようとするが,過剰な小胞体ストレス下ではアポトーシスが誘導される.UPRにはinositol-requiring enzyme 1α(IRE1α),PKR-like ER kinase(PERK),activating transcription factor 6(ATF6)の3経路があり,小胞体ストレスからの回復からアポトーシス誘導までを担っている48, 49).
Sarnyaiらは,ラット膵島細胞RIN-5FをパルミトイルCoAの材料となるパルミチン酸(C16:0脂肪酸)で処理すると,C16:0-セラミドとC18:0-セラミドが増加し小胞体ストレスが惹起されることを報告している50).また,Choiらは,腸管上皮細胞へのミリスチン酸(C14:0脂肪酸)処理では,CERS5/6の作用によりC14:0-セラミドが増加して小胞体ストレスが誘導されることを報告した51).他方,CERSが合成するセラミド分子種の違いによって小胞体ストレスへの反応が異なる現象がみられている.たとえば,ヒト頭頸部扁平上皮がん細胞においてはCERS1が産生するC18-セラミド(C18:0-およびC18:1-セラミド)とCERS6が産生するC16:0-セラミドでは,小胞体ストレスに対して異なる反応を示すことが報告されている52).さらに,ツニカマイシンやヒストン脱アセチル酵素阻害剤スベロイルアニリドヒドロキサム酸処理での小胞体ストレス誘導時には,CERS6とC16:0-セラミドの減少が,小胞体のカルシウムイオン(Ca2+)ポンプsarco/endoplasmic reticulum Ca2+-ATPase 2(SERCA2)およびSERCA3の発現・活性の低下を引き起こして小胞体からのCa2+放出を促進する53).その結果,小胞体-ゴルジ体膜のネットワークが破壊され,ゴルジ体が断片化することがきっかけとなり,ATF6が活性化してERストレスからアポトーシスが誘導される53).このことから,CERS6–C16:0-セラミドが小胞体ストレスに対して保護的に働いていることが示唆される.一方,ヒト肝がん細胞Hep3Bへのパルミチン酸処理による小胞体ストレスではCERS6の過剰発現は小胞体ストレスを悪化させる54).興味深いことに,CERS6と同様にC16:0-セラミドを増加するCERS5過剰発現は小胞体ストレスに影響を与えなかった.このことから,CERS5とCERS6の合成するC16:0-セラミドには異なる性質があると考えられるが,そのメカニズムは不明である.また,この報告では,極長鎖セラミド(C22–C24-セラミド)の処理あるいは極長鎖セラミドを産生するCERS2の過剰発現は保護的に働いた.HammerschmidtらのグループはCERS6により合成されるC16:0-セラミドは,GRP78/BiPに結合することで小胞体ストレス誘導を促進するということを報告した55).他方,Liuらのグループは,腺様嚢胞がん細胞へのC2:0-セラミド処理によって,SERCAの抑制により小胞体のCa2+が放出し,IRE1αとPERKを介した小胞体ストレスからアポトーシスが誘導されることを示した56).CERSの阻害剤であるフモニシンB1の処理によって,C2:0-セラミドによる小胞体ストレスが抑制されることから,SERCAの抑制はC2:0-セラミドによる直接の作用ではなく,サルベージ経路を介した内在セラミド増加が関わっていると考えられる56).さらに,C2:0-セラミド処理による小胞体ストレスでは,UPRの中心制御分子であるシャベロンGRP78/BiPの発現も上昇することが示されている56).
これらの培養細胞レベルで見られるde novo合成やサルベージ経路を介したセラミド産生による小胞体ストレス誘導は個体レベルでも確認されている.甲状腺機能亢進症ラットにおいて,C6:0-セラミドの投与やSPTを構成するサブユニットであるSPTLC1とSPTLC2の過剰発現によって視床下部腹内側核における小胞体ストレスの増加がみられる57).また,卵巣摘出ラットでは,SPT阻害剤ミリオシンの投与やSPTLC1のノックダウンによるSPTの発現・活性抑制によって,視床下部服内側核での小胞体ストレスが抑制される58).高脂肪食によって耐糖能低下とインスリン耐性が誘導されたマウスでは,C16–C22の長鎖セラミド量の増加とC22–C24の極長鎖セラミドおよびSM量の減少が引き起こされるなど,小胞体ストレスとの関連が見いだされている59).
以上の報告ではセラミド産生により小胞体ストレスが惹起されていたが,逆に小胞体ストレスを受けてセラミド産生が誘導されることも報告されている.初代ヒト膠芽腫細胞では,PERKの阻害によってセラミド産生が抑制される60).melanoma differentiation-associated gene-7/interleukin-24(MDA-7/IL-24)の過剰発現を利用した初代ヒト膠芽腫細胞の細胞死誘導では,小胞体ストレスシグナルが活性化されるが,PERKの阻害によってセラミド増加とCa2+のサイトゾルへの流入,ROSの産生が抑制される60, 61).逆に,ミリオシンによるde novo合成阻害やCERS6のノックダウンによって,MDA-7/IL-24によるCa2+上昇とROS産生が止まる60).また,aSMase経路でも同様のことが起きており,PERKがCERS6やaSMaseの上流でUPRを制御していることが示唆されている60).小胞体で合成されたセラミドはCERTによってゴルジ体へ輸送されてSM合成へ利用されるが,SMS阻害剤であるD609処理によって,小胞体セラミドが蓄積してオートファジー関連細胞死が誘導されることが報告されている62).近年,小胞体のde novo合成経路やサルベージ経路のセラミド産生に関わる酵素が転写レベルで制御されることで,「セラミドホメオスタシス」が維持されており,この崩れによってセラミドを介した細胞死や細胞老化が起こることが報告されている63, 64).
3)ゴルジ体セラミド
先に述べたようにゴルジ体では,小胞体から輸送されたセラミドを元にSMやGSLs, C1Pなどが合成される.現在,セラミド産生に焦点を絞ると,先にあげた複合スフィンゴ脂質の合成酵素の阻害によってゴルジ体にセラミドが蓄積するようなことは報告されていない.しかしながら,nSMase2によるSM加水分解でゴルジ体セラミドが蓄積することが報告されている65).Backらのグループは,ラット神経膠芽腫PC-12細胞の栄養飢餓時のオートファジーにおいて,ゴルジ体のnSMase2がセラミドを産生することでp38 MAPKのリン酸化とmammalian target of rapamycin(mTOR)の抑制を介するオートファジーを誘導し,細胞の生存率を増加させることを報告している65).
4)形質膜セラミド
アポトーシス経路には先ほど述べた内因性経路の他に細胞死受容体(death receptor)を介した外因性経路がある66, 67).最終的には外因性経路もカスパーゼ-9やカスパーゼ-3の活性化を経る内因性経路と同様のプロセスをたどるが,外因性経路はサイトゾルでのカスパーゼ-8活性化が指標となる.細胞死受容体にはFas receptor(Fas),TNF-receptor(TNFR),TNF-related apoptosis-inducing ligand receptor(TRAILR)などが知られており,それぞれの細胞外リガンド[Fas-L(CD95L),TNF-α, TRAILなど]が結合すると,形質膜上の受容体が細胞死誘導性シグナル伝達複合体(death-inducing signaling complex:DISC)を形成し,カスパーゼ-8を介した外因性経路を誘導する67).この形質膜から起こる外因性経路の活性化にもセラミド産生が関わっている.CD95LやTRAILの刺激によってリソソームから分泌されたS-aSMaseが形質膜のSM加水分解を経てセラミド産生を誘導し,セラミドの偏在した微小ドメイン(ceramide-rich platforms:CRPs)を形成し,FasやTRAILRのクラスター形成を促すことが報告されている61, 68).また,Jurkat T細胞への深紫外線(ultraviolet-C:UV-C)処理によっても,S-aSMaseがCRPsを形成して,FasによるFADDやカスパーゼ-8活性化の場所を提供することが示されている69).このCRPsを介したクラスター形成は,TRAILとdeath receptor 5(DR5, TRAILR2)間でも確認されており,アポトーシスを誘導している70).また,メラノーマや頭頸部扁平上皮がん細胞などのさまざまながん種の細胞において,抗がん剤であるリン酸化酵素阻害剤ソラフェニブとヒストン脱アセチル化酵素阻害剤ボリノスタットの併用によってaSMaseの活性化とde novo合成が誘導され,CD95とその受容体がCRPsでクラスター化して下流のPERKを活性化することで小胞体ストレスからのアポトーシスやオートファジーを誘導することが示されている71, 72).
HL-60細胞がTNF-α刺激によって単球へと分化する際にはnSMaseが活性化されることが1991年に発見された73).その後,TNFRのサイトゾル側でfactor associated with neutral sphinogomyelinase(FAN),receptor for activated C-kinase(RACK1),polycomb protein embryonic ectoderm development(EED)が形質膜nSMase2と複合体を形成し,セラミドが産生されることが示された25).また,他のサイトカインIL-1βやIFN-γ刺激に対しても形質膜nSMase2の活性化を介したセラミド産生が誘導されており炎症応答に関与している74, 75).他方,形質膜nSMase2は好中球の食作用にも関連している.細菌の貪食中に形質膜でnSMase2は活性化するが,nSMase阻害剤GW4869により好中球の運動や遊走が抑制される76, 77).形質膜でのnSMase2活性化からセラミド増加による炎症や貪食作用では,セラミドの直接作用だけでなく,後述するceramide-activated protein phosphatases(CAPPs)を介したシグナル伝達や他の複合スフィンゴ脂質への変換なども関与している78, 79).また,ヒト子宮頸がん細胞株HeLaにおける致死量未満の抗がん剤ドキソルビシンとボリノスタチン処理では,nSMase2の活性化によって形質膜でのセラミドが増加し,細胞遊走や細胞接着に関わる分子を制御することが報告されている80).他方,Wonらのグループは,マウスの線条体およびラット神経膠芽腫PC-12細胞でのドーパミントランスポーターを介したドーパミンの細胞内への取り込みに,形質膜でのnSMase2を介したセラミド産生が重要であることを報告している81).
5)リソソームセラミド
リソソーム経路を介したアポトーシスでは,リソソーム膜の膜透過性亢進によってカテプシンがプロセシングを受けて活性化された後に放出され,切断型Bid(tBid)を介した内因性アポトーシス経路を経て細胞死が誘導される66).TNF-α刺激によるカテプシンDを介したアポトーシスでは,TNF-αとTNFRが細胞内にエンドサイトーシスで取り込まれてリソソームへ輸送される過程で,カスパーゼ-8がカスパーゼ-7を活性化する82).その後,活性化カスパーゼ7がプロセシングにより不活性型のaSMaseを活性化することでリソソームセラミドが上昇し,その結果,カテプシンDが活性化・放出すると報告されている82–84).また,ヒトナチュラルキラー由来がん細胞KHYG-1において,増殖に必須なインターロイキン2(IL-2)を除去すると,aSMaseの活性化によるリソソームでのセラミド蓄積,カテプシンBの活性化・放出,外因性アポトーシスの誘導が起こることが報告されている85).カテプシンDとセラミドは直接相互作用する83)が,カテプシンBとセラミドの相互作用は不明である.むしろ,リソソーム膜でのセラミド上昇がリソソーム膜ダイナミクスに関与している可能性がある85).また,先に紹介した小胞体ストレスに対してもリソソームにおけるセラミド蓄積が関わることが示されている.リソソームに局在するaCDaseの阻害剤LCL521の処理によってリソソームでC16:0-セラミドが蓄積し,カテプシンBとカテプシンDの両方が活性化され,小胞体ストレスの増加からオートファジーが誘導されることが報告されている86).また,ヒト神経膠腫のオートファジー誘導でもaSMaseの活性化からカテプシンBが活性化・放出されて細胞死が誘導される87, 88)
6)エンドソームセラミド・エクソソームセラミド
先に述べたように,エンドソームのスフィンゴ脂質組成の変化が物質輸送の制御を介して細胞の運命を変えることが報告されている24).他方,エクソソームを含むEVsの放出では,エンドソームにおけるnSMase2によるセラミド産生が重要であることが報告されている25).また,抗がん剤耐性HL-60/ADR細胞では,エクソソームのセラミド割合が増えることでその放出が増え,それが抗がん剤耐性能の獲得に関連することが報告されている89).エクソソームを含むEVs産生は,がんだけでなくアルツハイマー病やパーキンソン病など神経変性疾患においても重要であるため,nSMase2を介したセラミド産生もこれらの疾患に関与するだけでなく治療標的にもなりうる90).EVsにおけるセラミドの機能については湯山の稿に記載されている.
7)核内セラミド
ヒトのリンパ芽球細胞において,抗がん剤ネオカルチノスタチンがDNA二本鎖切断(DNA double-strand breaks:DSBs)を介してアポトーシスを誘導する際に,核内のnSMaseの活性化によってセラミドが増加することが報告されている29).DSBsによるアポトーシス誘導にはataxia-telangiectasia mutated(ATM)の活性化が重要で,毛細血管拡張性運動失調症患者由来のATM欠損リンパ芽球細胞では,ネオカルチノスタチン処理によってセラミドが増加するにもかかわらず細胞死が誘導されない.このため,セラミドがATM活性化に関わっていることが示唆されている29).また,HL-60細胞への抗がん剤アドリアマイシン処置やヒトJurkat T細胞へのFas処理では,核内nSMase活性化を介したセラミドの増加を伴うアポトーシスが誘導される91).同様に,Albiらのグループは,UV-C照射によってラット濾胞性甲状腺FRTL-5細胞においてnSMaseを介した核内セラミド上昇を伴うアポトーシスが誘導されることを報告した33).しかしながら,核内のnSMaseの実態やセラミドによるATM活性化を含め,核内から誘導されるアポトーシスにどのようにセラミドが関与するかについては不明な点が多く残されているため,今後の研究が期待される.
8)その他のセラミド標的分子
セラミドが標的として活性化するタンパク質脱リン酸化酵素の総称としてCAPPsが知られる92).先に述べたHL-60細胞におけるTNF-α刺激時には,SM加水分解によってセラミドが増加し,CAPPsの活性化を通じてc-mycが抑制され,細胞増殖が抑えられる93).CAPPsとしては,protein phosphatase 1(PP1)およびprotein phosphatase 2A(PP2A)がよく知られており,セリン/トレオニン脱リン酸化酵素に属する.セラミドによって活性化されたPP1はmRNAのプロセシングタンパク質であるserine/arginine-rich protein splicing factor(SR)を脱リン酸化し,カスパーゼ-9とBcl-xLの選択的スプライシングを制御することで内因性アポトーシス経路を活性化させることが報告されている94).また,ヒト肝細胞がんHepG2細胞においても,セラミドが活性化したPP1によるBcl-xLのスプライシング制御がS-アデノシルメチオニン誘導性のアポトーシスに関与している95).同様に,Limらはエタノールによる神経細胞死において,de novo合成を介したセラミドの増加がPP1を活性化してSRを抑制し,myeloid cell leukemia 1(MCL-1)の異常なスプライシングフォームMCL-1Sを産生することで,小胞体–ミトコンドリア相互作用を増強し,ミトコンドリアからのCa2+流出,ROSを経てアポトーシスを誘導することを報告した96).セラミドによって活性化されたPP2Aは,c-mycに加え,PKC97),c-Jun98),Bcl-299)を抑制し,内因性アポトーシスを誘導することが報告されている.特に,セラミド誘導性のPP2AによるBcl-2の脱リン酸化では,Bcl-2/Bax複合体からBcl-2が遊離し,BaxがMOMPを引き起こすと同時に,遊離したBcl-2がp53などと結合してアポトーシスを促進する99).上述のさまざまながん種でのソラフェニブとボリノスタットの併用では,CD95を介した小胞体ストレスにおいてPP2Aを介したROSの産生が起こる100).最近,Liaoらのグループは,赤血球の増殖・分化では転写因子GATA-1によるセラミドのde novo合成酵素の包括的制御による「セラミドホメオスタシス」が維持されているが,その異常によるセラミド蓄積がPP2Aを活性化し,AktやERK MAPK, STAT5を脱リン酸化してアポトーシスを誘導することを報告した63).他方,栄養飢餓条件下ではaSMaseの活性化によるセラミドの増加が,CAPPsを介してmTORを脱リン酸化し,不活化することでオートファジーおよびオートファジー関連細胞死を誘導するということが報告されている101).また,PP2Aを介したmTORの抑制は制御性T細胞の機能と自己免疫抑制にも関与している100).他にも,B16-F10メラノーマ細胞への放射線照射ではaSMaseの過剰発現によってオートファジーを介した細胞死が促進すること102),肝細胞がんへのメラトニン誘導性の細胞死ではaSMase–セラミド増加がオートファジーによる細胞死を引き起こすことが報告された103).セラミドが活性化するPP2Aの標的にはアクチン骨格再編成に関わる分子もある.ヒト乳がんMCF7細胞へのシスプラチン処理では,S-aSMaseの活性化によってセラミドが産生し,PP2Aが活性化される.活性化したPP2Aは足場タンパク質であるEzrinを脱リン酸化して局在変化を誘導することで,形質膜への局在ができなくなり,アクチン骨格再編成が抑えられて細胞遊走が阻害される104).
以上,セラミドが細胞死や分化,遊走,オートファジーなどさまざまな細胞生理作用を引き起こすことが報告され,その標的分子も徐々に明らかになりつつある.