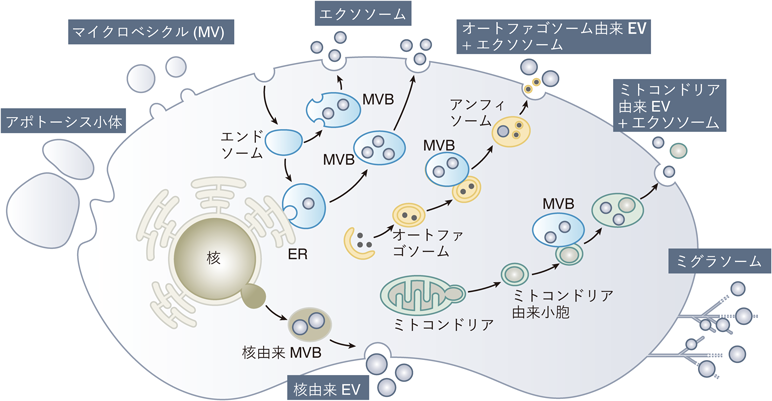
細胞外小胞(extracellular vesicle:EV)は,ほぼすべての真核細胞から産生されるナノスケールの脂質二重層膜小胞である1).EVには生成機構の違いによって多様なタイプが存在する(図1).その中でよく知られているのは,エンドソーム由来のエクソソーム,形質膜から出芽してできるマイクロベシクル(microvesicle:MV),アポトーシス過程で生じるアポトーシス小体である.エクソソームの生成過程は以下のとおりである.エンドソーム膜の一部が内側に陥入し,内腔小胞(intraluminal vesicle:ILV)が形成される.これにより多胞体(multivesicular body:MVB)が生成される.MVBの一部はリソソームと融合し分解されるが,残りの一部は形質膜と融合してILVを細胞外に放出する.この放出されたILVがエクソソームと呼ばれる2).最近の研究では,エンドソームから独自にMVBが生成される一般的な経路の他に,エンドソームと小胞体(ER)が相互作用してMVBを形成する機構も報告されている3, 4).また,オートファジー過程で形成されるオートファゴソームやミトコンドリア由来の小胞(mitochondria-derived vesicle:MDV),核膜(nuclear envelope:NE)由来の小胞からもEVが生成されることも確認されている5–7).これらオートファゴソームやMDVは,MVBと融合することでエクソソームとともに細胞外に放出される.当初EVは細胞内の不要物質を排出する役割を持つと考えられていたが,現在ではEVが持つ多様な機能が明らかになっている.特にエクソソームに関しては,RNA,タンパク質,脂質,糖鎖などのさまざまな分子を特定のターゲット細胞に運搬し,細胞間の分子輸送に関与することが多くの研究で示されている(総説8, 9)参照).
セラミドは,スフィンゴイド塩基と脂肪酸がアミド結合して形成される脂質分子である.また,セラミドを基本骨格として,その頭部にホスホコリンが結合したスフィンゴミエリン(sphingomyelin:SM)や糖鎖が結合したスフィンゴ糖脂質(glycosphingolipid:GSL)が生成される10).これらのセラミドや複合スフィンゴ脂質は,形質膜の構成成分であるだけでなく,エクソソームやMVなどのEVにも豊富に含まれている.なぜこれらの脂質分子がEVに含まれるのか,本稿ではEVの生成過程や機能発現においてスフィンゴ脂質が果たす役割について紹介する.
これまでのEVのリピドミクス解析の報告は,少数の細胞種のエクソソームとMVに関するもののみで,EVの脂質組成についての知識はまだ限定的である.EVはドナー細胞の膜由来であるため,形質膜の構成脂質がEVにも検出される.しかし,興味深いことに,これらEVの脂質組成はドナー細胞のものと異なり,セラミドやその代謝脂質であるSM, GSLの濃度がドナー細胞より高いことが複数の研究で報告されている11–18).これらの濃縮率は細胞種によって異なる.たとえばエクソソームにおいて,SMは,マウスオリゴデンドロサイト様細胞Oli-neu細胞由来では親細胞の1.5倍に13),ヒト前立腺がん由来細胞PC-3細胞由来では2.4倍15),ヒトB細胞由来では2.3倍17),肥満細胞RBL-2H3由来では2.8倍12),マウス神経芽細胞腫Neuro2a細胞由来では2.2倍18),樹状細胞由来では2.2倍11)に濃縮されている.また,MVのSMに関しては,骨髄間葉系幹細胞(MSC)由来では2倍,ヒト肝臓がん細胞Huh7細胞由来では2倍19),血小板由来では3.4倍20)である.
GSLに関してはエクソソームの報告のみだが,その濃縮率には大きなばらつきがある.たとえば,Oli-neu細胞由来エクソソームではGSLが親細胞の2.0倍,PC-3細胞由来では3.8倍,マウス神経芽細胞腫Neuro2a細胞由来では18倍に濃縮されている18).エクソソームに豊富なSMとGSLは,形質膜の脂質マイクロドメインに特徴的な成分である.他の脂質マイクロドメイン成分であるコレステロールの濃度も親細胞に比べて高いため,エクソソームは脂質マイクロドメインのような秩序液晶相に富む膜で構成された小胞であると考えられる.
エクソソーム中のSMとGSLは,すべての報告でドナー細胞より高濃度であるが,セラミドに関してはばらつきがみられる.PC-3細胞由来のエクソソームではセラミドが1.3倍15),Oli-neu細胞由来では3.3倍13),Neuro2a細胞由来では12.3倍18),骨髄MSC由来MVでは2倍,Huh7細胞由来MVでは2.4倍19),血小板由来MVでは3.4倍20)と増加している.しかし骨髄MSCやU87細胞由来のエクソソームでは変化がない19).また逆にHuh7由来エクソソームでは0.25倍とセラミド量は低くなっている19).このセラミドの含有率の差異は,そのEVの産生過程のセラミド依存度によるのかもしれない.またEV膜においてセラミドがどのように分布しているのか,その存在様式は未解明だが,セラミドリッチプラットフォーム(ceramide-rich platform:CRP)と呼ばれる新規の脂質マイクロドメインの存在も提唱されている21).CRPでは,セラミドがセラミド結合タンパク質との相互作用を介して,EVカーゴ分子の積み込みを制御している可能性も示唆されている.
さらに,刺激に応じて,EVのセラミド含有率が変動する例も報告されている.乏突起膠腫細胞株をサイトカイン(TNF-αとIFN-γ)で処理すると,セラミド(C16:0-,C24:0-,C24:1-セラミドとジヒドロセラミド)に富むEVの形成と放出が誘導されることが示されている22).また,胚性海馬細胞をビタミンD3で処理すると,EV中のセラミド含量が増加することも報告されている23).しかし,セラミド高含有EVの形成機構は現在のところ不明である.
1)形質膜からのマイクロベシクル形成
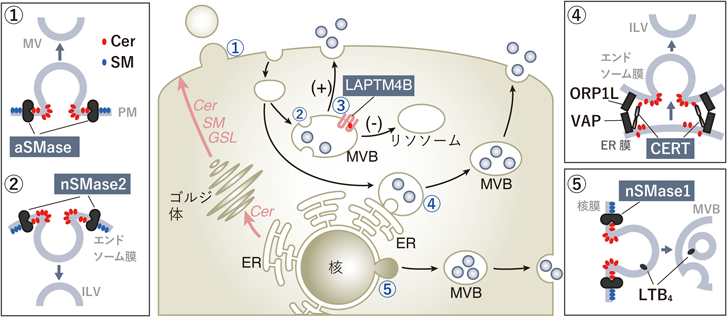
セラミドはERで新規に生合成され,その後ゴルジ体に輸送される.そこでSMやGSLに変換され,細胞膜に輸送される.また,SMはスフィンゴミエリン分解酵素(SMase)の作用によって加水分解され,セラミドに再変換される.SMaseのうち酸性SMase(aSMase, SMPD1)は,主にリソソームでのSM分解を担うとされているが,存在する部位の膜脂質組成によって酵素のKm値が変化するため,中性のpH下でも酵素活性を維持することが可能であると報告されている24).実際,骨髄細胞や血管内皮,赤血球では,特定の刺激により分泌型リソソームと形質膜が融合し,aSMaseが細胞外に放出され,形質膜外層のSMに作用してセラミドが生成される25).このaSMase依存的なセラミド生成によって形質膜の突出とそれに続くMVの放出が引き起こされることが知られている(図2-①).aSMase依存的なMV放出は,ストレス条件下の細胞(たとえば,鎌状赤血球症の赤血球)や危険シグナル(たとえば,ATP)などに刺激された細胞などで観察される26).
セラミドによるMV形成のメカニズムはまだ明確ではないが,以下の三つの可能性が指摘されている.(1)セラミドは円錐状の構造を持ち,膜に自発的な湾曲を与えることができる.この性質は,形質膜外層で生成されたセラミドが内層に再分配されることと組み合わされ,構造的な膜突出につながる可能性がある27, 28).(2)コレステロールと親和性の高いSMの分解は,形質膜上のコレステロールの拡散を促進し,流動性を高めることで,膜の突出を誘導する29).(3)セラミドが他の脂質から分離し,セラミド濃縮ドメインを形成することで,形質膜の外側への突出が起こる可能性も考えられる.SMを含む人工単分子膜をSMaseで処理すると,線張力と双極子静電反発によって制御される形状を持つセラミドに富むマイクロドメインが形成されることが観察されている30).
2)エンドソームからのMVB形成とエクソソーム放出
形質膜に存在するセラミドや関連脂質は,エンドサイトーシスによってエンドソームの膜成分として細胞内に再取り込みされる.エンドソームからMVBが形成される過程にもセラミドが関与していることが知られている.この過程では,中性スフィンゴミエリナーゼ2(nSMase2, SMPD3)が重要な機能を担っており,エンドソーム膜上でSMをセラミドに変換することによりMVB形成(ILV形成)を誘導する(図2-②)13).ILV形成は通常,細胞分裂過程などでも働く膜リモデリング機構,特にendosomal sorting complex required for transport(ESCRT)機構に依存していることが知られている31).ESCRT機構では,四つのタンパク質複合体(ESCRT-0,-I,-II,-III)が,エンドソーム膜のサイトゾル側の表面に結合し,膜の内側への出芽と切断を促進してILVを形成する.
一方,セラミドはESCRT機構に依存しない方法でもILV形成を促すことが可能である.SMを含むリポソームにnSMaseを作用させる実験では,SMのセラミドへの分解により,セラミド濃縮ドメインが形成され,膜が内腔側に屈曲し,最終的に小胞が生成されることが観察されている13).ESCRT依存的およびセラミド依存的なMVB形成機構は,同一の細胞内でも共存しており,細胞の種類によってそのバランスが異なる.さらに,極性上皮細胞を用いた実験において,SMase依存的なセラミド産生機構が基底外側からのエクソソーム放出を制御し,一方でESCRTタンパク質依存的な機構(この場合はAlix-Syntenin 1-Syndecan 1機構)が頂端側からのエクソソーム放出を制御する現象も報告されている32).これらの極性エクソソームは積荷タンパク質が異なるため,細胞は異なるカーゴ分子を持つエクソソームを産生するために,複数のエクソソーム産生装置を使い分けていると考えられる.
また,逆にセラミドからSMを合成するスフィンゴミエリン合成酵素2(SMS2)の発現低下がMVB形成を誘導し,エクソソーム放出を促進することも示されている33).SMS2はエンドソーム膜にも発現しており,SMaseとは逆に,SMS2活性によりエンドソーム膜のセラミドを減少させ,MVB形成(エクソソーム放出)を抑制すると考えられる.また,SMS2は,ミグラソーム(migrasome)と呼ばれる小胞の産生に関与していることも報告されている34).ミグラソームは,遊走細胞が移動に伴って細胞の後端に形成するリトラクションファイバーという細長い突起において形質膜から産生される小胞である(図1).成熟すると自然破裂や漏出によりシグナル分子などを放出するが,一部は小胞のままEVとして分泌されることが知られている.SMS2はミグラソーム産生に必須のタンパク質で,SMS2の集合体が存在する部位において,セラミドがSMに変換されることでミグラソーム形成が誘導される.またミグラソーム形成には,長鎖セラミドの合成に必要なセラミド合成酵素CerS5と,セラミド輸送タンパク質(ceramide transporter:CERT)が関与することも報告されている34).
3)LAPTM4BによるMVB輸送制御
LAPTM4B(lysosomal protein transmembrane 4B)は,主にリソソームと後期エンドソームに局在する4回膜貫通タンパク質であり,その第3膜貫通らせん構造内にスフィンゴ脂質結合モチーフを持っている35, 36).セラミド,特にC16やC18などの長鎖脂肪酸で構成されるセラミドで神経細胞株を処理すると,EVの放出が促進され,このEV放出がLAPTM4B遺伝子のノックダウンにより抑制される37).タンパク質–脂質オーバーレイアッセイを用いて,長鎖脂肪酸含有セラミドがLAPTM4Bに結合することも確認されている.外部から添加された長鎖脂肪酸含有セラミドは細胞に取り込まれ,後期エンドソームやリソソームに滞留し,そこでLAPTM4Bと相互作用してEV産生を促進すると考えられる(図2-③).MVBの蛍光イメージング解析によれば,セラミド処理後にはLAPTM4B依存的にMVBのリソソームへの輸送が抑制され,逆に形質膜への輸送が促進される現象が観察されており,これがEV放出の増加につながると考えられる.さらに,LAPTM4Bを欠損した細胞では,放出されるEVの脂質組成,特にスフィンゴ糖脂質の組成が変化することが報告されており,LAPTM4Bがスフィンゴ糖脂質のEVへのソーティング機能を持つ可能性もある38).
動物細胞におけるLAPTM4B依存性のEV放出が,外因性の植物型セラミドによっても誘導されることが報告されている39).セラミドを構成するスフィンゴイド塩基の化学構造は生物種によって異なり,動物ではスフィンゴシン(trans-4-スフィンゲニン,d18:1)が一般的であるが,植物のセラミドは主に4,8-スフィンガジエニン(d18:2)で構成されている(詳細は石川の稿および菅原の稿を参照).化学合成されたd18:2型セラミドで神経細胞株を処理した場合,d18:1型セラミド処理した場合と同様に,LAPTM4B依存性のEV放出の誘導が観察される.d18:2型セラミドでも,C16やC18のような長鎖脂肪酸を含むタイプのセラミドがEV産生の誘導能が高いことが示されている.これらの研究結果を踏まえると,セラミドやその誘導体を用いたEV放出の制御に関する開発研究が可能であることが示唆される.
4)CERTによるERからのセラミド輸送とMVB形成
セラミド輸送タンパク質CERTは,SMの生合成に必須のタンパク質である40).セラミドがERで合成された後,CERTはER膜からセラミドを抜き出し,ERと遠位ゴルジ体の膜接触部位(membrane contact site:MCS)を経由してゴルジ体膜へと輸送する41, 42)(花田の稿を参照).ゴルジ体においては,受け渡されたセラミドから細胞機能に必要なSMが合成される.2011年の研究では,CERTが,ゴルジ体だけでなく,性感染症の原因となるクラミジア・トラコマティス(Chlamydia trachomatis)が宿主細胞内に作る封入体にも,ER–封入体のMCSを通じてセラミドを輸送することが示されている43)(詳細は花田の稿を参照).この寄生細菌は,宿主細胞が合成したセラミドを自己の封入体に取り込んでスフィンゴミエリンに変換することで増殖することが可能になる.
さらに近年の研究では,CERTが,ERからエンドソームへセラミドを受け渡し,MVB形成(ILV形成)とEV放出を引き起こすことが明らかになった3, 4).エンドソーム上のオキシステロール結合タンパク質関連タンパク質1L[oxysterol binding protein(OSBP)-related protein 1L:ORP1L]は,エンドソーム膜のコレステロールレベルに応じて構造が変化することで,ERに常在するアダプタータンパク質であるvesicle-associated membrane protein-associated protein(VAP)との相互作用が促進され,これにより,ERとエンドソームのMCSが強化される44).VAPはER膜上でCERTとも結合しており,MCSにおいてCERTがERのセラミドをエンドソーム膜側へ受け渡し,エンドソーム膜にセラミドが蓄積することでMVB形成(ILV形成)が引き起こされる(図2-④)4).さらに,ER-エンドソームMCSにおいて,ORP1LはMVBの分解経路への輸送に関与する低分子GTPase Rab7aから,分泌性経路に関係する低分子GTPaseの一つであるArl8bへの切り替えを制御する45).このスイッチにより,Arl8b陽性エンドソームが形質膜へ移行し,EV放出が促進される.エンドソーム膜へ受け渡されたセラミドがILV形成を引き起こすメカニズムは明確ではないが,前述のエンドソームにおけるnSMase依存的なILV形成と類似した機構が働いていると考えられる.また,CERTのエンドソーム膜への会合は,II型ホスファチジルイノシトール4-リン酸キナーゼα(PI4KII α)によって合成されたPI4PとCERTのプレクストリン相同(pleckstrin-homology:PH)ドメインとの間の結合によって起こることが報告されている3).
細胞間でのRNA輸送は,EVの重要な機能の一つであるが,EV内へのRNAの選択的なソーティングのメカニズムはまだ十分に理解されていない.VAPやCERTをノックダウンすると,EV内の特定のRNAの量が減少することが報告されており,これはCERT関連機構がセラミドの移動だけでなく,EVへのRNAの積み込みにも関与している可能性を示唆している4).
5)核膜からのMVB形成
2022年,Aryaらによる好中球を使用した実験報告では,エンドソームではなく核膜からMVBが形成され,直径約300 nmの核膜由来のEVが放出されることが明らかになった7)(図2-⑤).好中球は,損傷を受けた組織へ効果的に浸潤するため,ロイコトリエンB4(LTB4)を分泌し,これを周囲の好中球への障害シグナルとして伝達する.このLTB4とその合成酵素群(5-lipoxygenase, lipoxygenase activating protein, leukotriene A4 hydrolase)はEVにパッケージされて放出されることが報告されている46).これらのLTB4を含むEVは,標準的なエンドソーム由来のMVBではなく,LTB4やその合成酵素が局在する核膜由来のMVBから放出されることが明らかになっている7).核膜(内膜)では,中性スフィンゴミエリナーゼ1(nSMase1, SMPD2)の作用によりSMからセラミドが生成し,セラミド濃縮マイクロドメインが形成される.このマイクロドメイン依存的にLTB4がクラスター化し,核膜が細胞質側に湾曲して小胞が生成される.この核膜出芽小胞はその後,ILVを持つMVBに成熟し,このMVBが形質膜と融合することにより,LTB4陽性EVとして細胞外に放出される.核膜の出芽部位や核膜由来のMVBにはAlixなどのESCRTタンパク質が存在することから,核膜由来小胞のMVBへの成熟過程にESCRT機構が関与する可能性が指摘されているが,この過程にセラミドが関与している可能性もある.
6)セラミド代謝物の関与するEV産生
セラミドの分解産物スフィンゴシンがリン酸化されてできるスフィンゴシン1-リン酸(S1P)は,脂質メディエーターの一種で,形質膜上のS1P受容体(S1PR)に結合することにより,細胞運動,アクチン骨格の制御,細胞増殖などの多様な細胞応答を引き起こす47).HeLa細胞における実験では,セラミドとともにS1Pも,MVBの形成やILVへの分子のソーティングを誘導することが報告されている48, 49).エンドソーム膜でセラミドから代謝され生成されたS1Pは,同じくエンドソーム膜に存在するS1P受容体S1PR2を刺激し,Rho GTPaseの活性化とアクチン骨格の制御を介して,MVBの形成とCD63などのタンパク質やmicroRNA(miRNA)のILVへのソーティングを誘導する49, 50).また,S1Pは,HeLa細胞からアポトーシス過程で放出されるapoptotic exosome-like vesicle(AEV)の産生にも関与していることが報告されている51).このAEVはS1P/S1PR複合体を表面に保持しており,マクロファージがAEVを取り込むことにより炎症反応が惹起されることが示されている.
また別のセラミド代謝物であるガラクトシルスフィンゴシン[スフィンゴシンにガラクトースが結合した分子でサイコシン(psychosine)とも呼ばれる]が,オリゴデンドロサイトでのMVの産生に関与していることも報告されている52).ガラクトセレブロシダーゼ(GALC)遺伝子欠損によって引き起こされるリソソーム病の一種であるクラッベ病では,脳内のオリゴデンドロサイトにGALCの基質の一つであるサイコシンが徐々に蓄積し,これによってオリゴデンドロサイトが死滅し,脱ミエリンを引き起こす.クラッベ病のモデルマウスとして用いられるTwitcherマウスでの研究では,サイコシンの蓄積がMVの放出を促進することが示されている52).これは,蓄積したサイコシンが異常なMVの産生を誘導し,それが脱ミエリンに関与する可能性を示唆している.
EVに組み込まれたセラミドやその代謝物の放出がドナー細胞にとってどのような意義を持つのか,また,これらがターゲット細胞にどのように作用するのかは重要な問いである.しかし,現在のところ,培養細胞を用いた実験報告が主で,明確な情報はまだ少ない.ここでは表1にまとめられた報告例のうち,アルツハイマー病病理形成の制御,リソソーム病におけるセラミド蓄積の緩和,単球系細胞の走化性誘導というEV含有スフィンゴ脂質の三つの機能について紹介する.
表1 スフィンゴ脂質が関与する細胞外小胞の生理的および病理的機能| EV含有スフィンゴ脂質 | EVドナー細胞 | EVターゲット細胞 | 機能 | 参考文献 |
|---|
| C16:0-セラミド,S1P | 肝細胞 | マクロファージ | 遊走促進,炎症反応 | 56 |
| ガングリオシド | ニューロン | ミクログリア | Aβ除去 | 18, 33, 55 |
| C18:0-セラミド | Aβ処理アストロサイト | アストロサイト | アポトーシス誘導 | 72 |
| C24:1-セラミド | (血液中) | 骨髄由来MSC | 老化促進 | 73 |
| C16:0-セラミド | 子癇性前置胎盤の絨毛細胞 | 内皮細胞 | 内皮細管形成阻害 | 74 |
| ガングリオシドGD2 | GD2発現メラノーマ | メラノーマ | 細胞増殖,浸潤,細胞接着 | 75 |
| ガングリオシドGD3 | GD3発現メラノーマ | メラノーマ | 遊走促進 | 76 |
| ガングリオシド | ニューロン | — | αシヌクレイン凝集 | 77 |
| セラミド | 胆道がん細胞 | 単球 | 炎症性サイトカイン放出 | 78 |
| S1P | パルミチン酸処理肝細胞 | マクロファージ | 遊走促進 | 79 |
1)EV含有スフィンゴ糖脂質とアミロイドβタンパク質代謝
筆者らの過去の研究により,神経細胞由来EV(特にエクソソーム)がアルツハイマー病の病因因子であるアミロイドβタンパク質(Aβ)と結合することが示された33).培養神経細胞から放出されるEVの脂質解析では,セラミド,SM,コレステロールに加え,GSLが由来細胞に比べEVに顕著に多いことがわかった18, 53).特に,ガングリオシドとして知られるシアル酸を含むGSL(GM1, GT1, GD1など)の含有率が高かった.以前の節でもふれたように,GSLはSMやコレステロールとともに脂質マイロドメインを形成する.このマイクロドメイン内で,GSL糖鎖は集積し,クラスターを形成するが,これにAβが結合することは以前から知られていた54, 55).神経細胞由来EV表面にもGSLクラスターが形成され,Aβが結合しやすくなっていると考えられる.Aβはアルツハイマー病の主要病理である老人斑の構成成分であり,老人斑にEVが局在しているとの報告も増えていることから56, 57),EVが老人斑形成に関与している可能性が示唆されている.我々の培養細胞やマウスモデルを用いた研究によると,EVは結合したAβをミクログリアに運搬し,分解することで細胞外のAβ蓄積(老人斑形成)を抑制することが示されている.これは神経細胞由来EVがAβ除去作用を持つことを意味する.さらに,我々が開発したAβ結合型GM1含有EVの高感度検出システムを用いた解析によると,アルツハイマー病モデルマウスの脳内でAβ蓄積が進むにつれて,血液中のAβ結合型GM1含有EVの増加が認められている58).これは,アルツハイマー病脳内では,EV依存性のAβ除去系が正常に機能していない可能性を示唆している.
2)EV依存的排出によるグルコシルセラミド細胞内蓄積の緩和
グルコシルセラミダーゼβ1(glucosylceramidase β1:GBA1)遺伝子の潜性変異は,リソソーム病の一種であるゴーシェ病を引き起こす59).GBA1は,グルコシルセラミド(GlcCer)をグルコースとセラミドに加水分解するリソソーム酵素をコードしている.GBA1の欠損は神経細胞でリソソームの機能不全とGlcCerレベルの上昇を引き起こす.ショウジョウバエを用いた実験により,神経細胞が活動する際にGlcCerが合成され,その一部がEVに含まれる形でグリア細胞へ輸送され,分解されることが示された60).また,同じモデルでの実験により,GlcCer含有EVの放出がグリア細胞のTGF-βによって誘導されることも明らかになった.これは,グリア細胞との連携を通じて,ニューロンがEVを放出しGlcCerの量を調整している可能性を示唆している.
GBA1変異はパーキンソン病のリスク因子でもあり,ゴーシェ病患者の家系内でパーキンソン病患者が頻出することが知られている61).また,GBA1の活性阻害は,パーキンソン病の病原因子であるαシヌクレインの蓄積や凝集を誘導することが,培養細胞系やモデルマウスを用いた実験で示されている62, 63).パーキンソン病患者由来のiPSニューロンでは,GlcCerの量とαシヌクレイン凝集体の量が正の相関を示し,GlcCer自体がαシヌクレイン凝集体の形成を誘導する可能性が指摘されている.リポソームを用いた実験では,GlcCerやGlcCerから脂肪酸が脱離したグルコシルスフィンゴシン[グルコサイコシン(glucopsychosine)とも呼ばれる]が,αシヌクレイン凝集体形成を促進することも示されている64).これらの事実を総合すると,ニューロンでのGlcCerの蓄積はパーキンソン病の病理形成の一因になると考えられる.そのため,EVを介したGlcCerの細胞外放出は,ゴーシェ病やパーキンソン病におけるαシヌクレイン蓄積を緩和する可能性があり注目されている.
3)EV含有セラミドと細胞遊走
EVに含まれるスフィンゴ脂質が細胞遊走を引き起こすことも報告されている.肝細胞をパルミチン酸(C16脂肪酸)で処理してERストレスを誘導すると,C16:0-セラミドを多く含むEVが放出される65).このセラミド高含有EVはマクロファージの走化性を活性化させることが示されている.マクロファージにおけるS1PのS1PR1への結合が走化性を促進することから,EVに含まれるS1Pが直接的に,またはEV内のスフィンゴ脂質がマクロファージに取り込まれた後S1Pに変換され,S1PR1への結合を介して遊走を誘導すると考えられる.非アルコール性脂肪肝炎(nonalcoholic steatohepatitis:NASH)患者の血液中エクソソームでは,C16:0セラミドとS1Pの含量の増加がみられる.肝臓組織への炎症性マクロファージの浸潤は,単純性脂肪肝からNASHへの遷移を引き起こす要因の一つである66).このNASHの重症化にEVに含まれるスフィンゴ脂質による遊走促進が関与している可能性があると指摘されている.