まずは,私(西道)の研究歴について書きたい.私は小学生のときから科学者になりたいと思っていた.そのために中学時代に「他の人とは違う生き方をする必要がある」と考えた.私は宮崎市在住だったので「より広い世界を知りたい」と考え,ロータリークラブが主催する交換留学生に応募した.中学3年のときは英語による集団面接で落第し,高校1年で再挑戦して採用され,高2の夏(1976年)より米国NY州Corinth Central High Schoolに1年間留学した.NY州といってもかなり北部の方だったので私が唯一の有色人種であったが,差別されることなどほとんどなく,むしろ少し成績がよかっただけで飛び級させていただき,正式な卒業証書(diploma)を授与された.ホストファミリー(Comings家:私の論文におけるミドルネームイニシャルCはComingsの略である)にも恵まれ,充実した1年を送った.そして,この経験は私の研究者人生に大きな影響を与えた.その後,大学院時代にも同じNY州のコーネル大学応用物理学科に留学し,論理的に英語で考え・話し・聴く能力を磨いた.米国では日本では経験できないようなストレスに遭遇し,これらに対応することによってメンタルを鍛えられた.一方で犠牲にしたこともある.日本の高校の3年生を経験しなかったため,数学では線形代数に関して,国語では百人一首に代表される古典の知識に関して後れを取ってしまった.
当時,海外の高校の卒業資格で受験できる国立大学は筑波大学と京都大学だけであった.私は,筑波大学の推薦入学制度により筑波大学が先に決まったため,筑波大学生物学類に進学した.当時の筑波大学は,学類(学部)を超えた授業を受けて単位を取ることができたため,私は可能な限り多くの講義(生物系・物理系・人文系)を受けた.寮に入って生活費が安かったので,4年間で約1000冊の本を購入し,大学4年間は,読書・勉強・運動(筑波ジョッギングクラブにて毎日約10キロ走った)・食事・睡眠だけに時間を費やした.人生の中で最も勉強した時間だった.その過程で,生物物理学教授の石坂昭三先生(故人)との運命的な出会いがあった.学部1年生のときから大学院のゼミに参加させていただき,とにかく生物物理学(物理学)を徹底的に勉強した.したがって,私の思考基板は物理学である.一方で,最終的に私は「物理だけでは生命はわからない」との結論に達し(シュレディンガーの波動方程式で解けるのはせいぜい水素原子で,より複雑な生命反応にはまったく歯が立たなかった.),その後,生化学(東京大学大学院薬学系研究科)と分子生物学[東京都臨床医学総合研究所(臨床研)]を学ぶこととなった.思い返せば,当時の生物物理学は後の革新的技術開発の基盤を作ったといえる.たとえば,FRET(fluorescence resonance energy transfer)の概念は1950年代に生まれていたが,実際に生命科学研究で使われたのは1990年代以降であった1).また,コーネル大学で私のデスクの横に座っていたWinfried Denkは後にベル研究所で2光子顕微鏡を発明し2, 3),今はマックス・プランク研究所の所長である.
ただし,20代の約10年間(学部後半・大学院・研究所)は,「他の人がやったことのない実験をしよう」との姿勢を貫き,やることなすことうまくいかなかった.しかし,この失敗歴のおかげで「どうしたら失敗するか」がわかり,「どうすれば失敗しないか」が予測できるようになった.30歳前後のことである.失敗し続ける私を温かく見守っていただいた大澤利昭先生(東京大学大学院薬学系研究科教授,故人)と鈴木紘一先生(臨床研部長,故人)にはあらためて御礼申し上げたい.約10年間失敗し続けたが,私はいずれ科学者として自立するという信念は揺るがなかった.この間,大学院と臨床研で同僚だった片桐康弘君(現NIH)には多くを教えていただいた.そして,30代に入ってようやく「狙った獲物は逃がさない」研究者になることができたように思う.
臨床研では,主にタンパク質分解酵素カルパインに関する研究を行った.その過程で,当時東京大学医科学研究所におられた大海忍先生の生化学会における「活性化カルパインを認識する抗体」に関する発表にインスピレーションを受け,「タンパク質の限定分解産物を認識する抗体の普遍的作製法」を確立することに成功した4–8).また,同僚の反町洋之君(その後,東京大学助手・東京都医学総合研究所グループリーダー,故人)に分子生物学の手ほどきを受け,この経験が理化学研究所(理研)におけるアルツハイマー病研究の分子生物学的・発生工学的研究の基盤となった.
カルパイン活性化を感度よく可視化する標的として,fodrin/spectrinの分解産物に対する抗体を作製した.当時臨床研部長であった月田承一郎部長(その後,京都大学教授,故人)からfodrin/spectrinの精製法を学んだ.この抗体は,兵庫医科大学脳神経外科の横田正幸助教授(当時)との共同研究に供され,脳虚血におけるカルパイン活性化を空間的に捕捉することに初めて成功した5).この抗体は多くの共同研究者に使われ,私が脳の疾患に関する研究を始めたきっかけとなった.また,アルツハイマー病研究で有名な丸山敬先生(現 埼玉医科大学教授)が臨床研に加わったことを契機として,脳虚血におけるアミロイド前駆体タンパク質(amyloid precursor protein:APP)を調べたところ,APPのβセクレターゼによるプロセッシングを促進することがわかった7).これは脳梗塞がアルツハイマー病のリスク因子であることと一致する.
また,大阪市立大学の森啓先生による,ヒト脳内に蓄積するアミロイドβペプチド(Aβ)が多くはAβ1–40で一部がAβ3pE-40(pE:pyroglutamate)であるとの報告9)があった.そこで,Aβ1-XとAβ3pE-Xに対する抗体を作製して高齢ヒト脳を病理学的・生化学的に解析した結果,我々は脳ではAβ3pE-42が大量に蓄積することを見いだした10–12).当初のAβの一次構造に関する研究でAβ3pE-42が見落とされた理由は,ペプチドの配列決定においてエドマン分解抵抗性があること,および,Aβ3pE-42は,通常のHPLCの条件ではカラムから溶出せず,カラムを50°Cに加熱し,塩基性の溶媒と低分子界面活性剤ベタインを用いる必要があったからである12).森啓先生の実験ではAβ3pE-42が精製途中で見落とされたが,pE特異的アミノペプチダーゼを利用することによって一部のAβ40がpE化していることを見いだしたという経緯があった.これがなければその後の展開はなかったであろう.製薬企業イーライリリーは我々の報告に注目し,Aβ3pE-Xに対するマウスモノクローナル抗体を作製し13),これをヒト化したドナネマブを作製した14).ドナネマブは,最近米国と日本で認可されたレカネマブと同様の効果を臨床試験にて示し,近く米国にて承認される模様である.レカネマブとドナネマブは標的エピトープが異なるので,両方を合わせたカクテル療法はより強い効果がみられると期待される.ただし,いずれの抗体も脳内に浮腫や出血を誘引するリスクがあるので,注意を有する.
上記の研究を行うにあたって井原康夫先生(東京大学神経病理学教授,故人)には大変お世話になった.今から30年近く前のことだ.私は自ら行った実験の結果を持って毎週のように臨床研のあった駒込から本郷まで自転車で通った.ディスカッションはしばしば2時間におよび,私はアルツハイマー病研究のイロハを学んだ.私の研究歴の中で夢のように楽しい季節であった.その後,井原先生は東京大学を退官されて同志社大学教授に着任されたが,次第に体調を壊され,入院療養されるに至った.2016年の5月28日に病床の井原先生からお電話があり,「私はもう終わりだから,日本の認知症研究をよろしく頼む」と伝えられ,私は大いに動揺した.その後持ち直されたので安心したが,私には忘れられない出来事であった.
研究上のストレッサー:不誠実な共同研究者.多くの研究者にとって最もストレスを感じる瞬間は投稿した論文がrejectされるときであるが,不誠実な共同研究者もストレッサーである.私が主要な役割を果たした仕事に関して私に論文原稿を見せずに無断で投稿した研究者がいる.Authorshipを都合よく決められたこともある.これは不正行為にあたるので,必要であれば論文取り下げを行うべきであろう.また,研究員を公募した際に虚偽の内容を含む推薦書を書かれ,研究室の運営に支障を来したことがある.私がこれまで共同研究を行ってきた研究者は軽く1000人を超える.中には約束を破った者がいたが,ほとんどは紳士的で,多くの友情を育んできたことは幸せなことである.
Ab3pE-42が脳内に蓄積する理由として,疎水性・凝集性が増すこと以外に分解抵抗性が生じることが考えられた.しかし,Aβが脳内でどのように分解されるかはまったくわかっていなかった.また,合成Aβペプチドを用いた試験管内の実験では分解酵素を絞り込むことは難しいと考えた.そこで,ラジオアイソトープで内部標識したAβ1–42を世界で初めて合成し,ラットおよびマウスの海馬に投与し,上記の条件のHPLCにシンチレーションカウンターをタンデムに連結して分解過程を解析する研究を立案した.その当時,理研脳科学総合研究センターが開設されるタイミングであったので,私は伊藤正男初代センター長(故人)に手紙を差し上げて研究計画をお伝えし,セミナーを行って2週間でチームリーダーとしての採用が決まった.偶然とはいえ,運がよかったと思う.当初標識Aβ1–42の合成は外注(New England Nuclear社)したが,数か月の後に担当技術者が突然「合成できない」とさじを投げた.我々は急遽ペプチド合成機を導入し,逐次合成を行った.ペプチド合成・精製・in vivo実験で活躍したのが岩田修永君(現 長崎大学教授)と津吹聡君(専門職研究員)の二人であった.我々は毎日のように実験が終わった後に2時間ほどディスカッションした.私が理研に着任したのが1997年秋で,実験室が使えるようになったのは翌年春であった.主要なAβ1–42の酵素がネプリライシンに代表される中性エンドペプチダーゼであるとの研究成果は2000年にNature Medicine15)と2001年にScience16)に発表され,伊藤正男センター長の期待に応えることができたことをうれしく思う.ちなみにAβ1–42の脳内半減期が約20分であるのに対してAβ3pE-42の半減期は70分以上である(Iwata et al., 投稿中17))ことから,後者が分解抵抗性を有することが判明した.
ところが,当時アルツハイマー病研究で最も権威のあったハーバード大学のDennis Selkoe教授は主要なAβ分解酵素はネプリライシンではなく,インスリン分解酵素(insulin-degrading enzyme:IDE)であると主張し18–20),国際学会で何度も対決することになった.IDEはミクログリアの培養上清より合成Aβの分解を指標に同定された分解酵素である.私はとにかく生データを披露して世界の研究者を納得させる必要を感じ,1か月かけて欧州と米国の主要な研究室を訪問し,合計10回のセミナーを行った.その過程で,ネプリライシンノックアウト(knock-out:KO)マウスとIDE KOマウスを使った実験により,ネプリライシンの優位が確定した.Selkoe教授は,しかし,フェアな研究者だった.大勢が確定したときの北米神経科学会で“Congratulations, sir,”というふうのことを言われ,私は驚いたと同時にうれしかった.私は米国人のこのような気質が好きだ.また,我々は2001年にネプリライシン遺伝子にリスク因子が存在することを予測した.これがgenome-wide association studies(GWAS)で実証されたのは2022年のことであった21).
その後,ネプリライシンの活性化因子としてソマトスタチンを同定した22).ソマトスタチンの脳内発現が加齢やADで低下していることとも一致する23, 24)しかし,ソマトスタチン受容体には5種類のサブタイプがあり,またホモ二量体とヘテロ二量体を形成することから,受容体の同定には時間を要した.最終的にサブタイプ1と4によって構成されるヘテロ二量体であるとの結論に達した25)(Nilsson et al., 投稿中26))(西道ら,国内特許7099717).また,II部で述べるように,綿村がソマトスタチン受容体を介したシグナル伝達にKATPチャンネルが関与することを見いだした.
APPの遺伝子変異が顕性遺伝形式を示す家族性アルツハイマー病(以下AD)の原因遺伝子として同定されて以来,多くのグループが遺伝子変異を有するAPP cDNAを過剰発現するトランスジェニック(Tg)マウスを作製した.代表的なものは,PDGFプロモーターを利用したPD-APPやプリオンプロモーターを利用したTg-2576, Thy1プロモーターを利用したAPP23である.本稿では過剰発現系を利用したモデルをTgと称する.その後,変異型APPとプレセニリン1(PSEN1)のcDNAの両方を過剰発現するダブルTgも多数開発された.プレセニリンはγセクレターゼの活性中心を担う.これらのモデルはAβ病理を再現するという点では成功したが,多くの問題があった(表1).そこで,我々は過剰発現によらずAD病理を再現する試みを開始した.その契機になったのが1999年にドイツのハイデルベルグ大学のBeyreutherグループによって発表されたPNAS論文である27).この論文では,培養細胞を用いた実験系で,APPのAβ(家族性ADの変異が複数見いだされていた)C末端前後のアミノ酸配列にphenylalanine scanによってアミノ酸置換を行い,Aβ42/Aβ40比に対する影響を検討していた.その結果,APPのI716F変異によりAβ42/Aβ40比が顕著に増加することを彼らは見いだした.私はデータを見たときに雷に打たれたような衝撃を受け,動物モデル作製に利用することを決めた.まさに,「神は細部に宿る」である.I716F変異はexperimental mutationであったが,約10年後にスペインのイベリア地方にて若年性家族性ADの変異としても同定された28)ことから,我々はこれをBeyreuther/Iberian mutationと名づけた.
表1 過剰発現モデルの問題点| 1 | 挿入されたcDNAによって内在性の遺伝子が破壊される.多くの場合にホモ接合体が胎生致死となる. |
| 2 | 本来APPを発現する細胞や細胞部位とは異なる位置に発現する. |
| 3 | 細胞選択的APP mRNAのスプライシングが失われる. |
| 4 | 過剰発現したAPPやPSEN1がさまざまな細胞因子と異常な相互作用をする.たとえばAPPとキネシンやJIP-1との相互作用に影響を与える. |
| 5 | 過剰に産生されたAPP断片は生理機能を有しており,これらが異常に作用する. |
| 6 | APPやPSEN1は膜貫通タンパク質であるため,Aβ病理とは無関係な小胞体ストレスを生じる. |
| 7 | 原因は不明であるが,Aβ病理とは無関係なカルパイン活性化を引き起こす(他のカルシウム依存性タンパク質も活性化される可能性がある). |
| 8 | AD脳とは異なるAβ分子種が蓄積される. |
| 9 | 明確な証拠はないが,トランスジーンに存在するプロモーターが転写因子のダイナミクスを撹乱する可能性がある. |
| 10 | セクレターゼ阻害剤の作用が本来のものと異なる場合がある. |
| 11 | 多くの場合に多数のラインを作製し異常(病理・行動)がみられたものが選択されてきたので,表現型バイアスが存在する. |
| 12 | 他の遺伝子改変動物と交配した際にアーティフィシャルな表現型が生じる場合がある. |
| 13 | マルチコピーのcDNAが挿入するため,ゲノム編集を利用することができない. |
| 14 | 病理が生じる前から行動異常がみられ,原因不明の突然死が生じる場合が多い. |
この当時は,ゲノム編集の技術がまだ確立されていなかったので,斉藤貴志研究員(後に副チームリーダーを経て現在名古屋市立大学医学部教授)が中心となって相同的組換えによってノックインマウスモデルを作製した.まず,マウスのAβをヒト化し,トータルAβの産生量を増加させるスウェーデン変異(NL)とI716F変異を導入したAppNL–Fラインを作製し,さらにAβの凝集を促進する北極変異(E694G)を導入したAppNL–G–Fラインを作製した.前者は9か月齢ごろからAβ蓄積が始まり18か月齢で顕著な病理が認められ,後者は4か月齢ごろから蓄積が始まり6か月齢を過ぎると顕著な病理が認められた.これらのマウスモデルはAD研究におけるgame changerと呼ばれ,現在国内外で800を超える研究グループによって使用されている.この次世代型ADモデルマウスを用いた研究によって,APP Tgの表現型の多くがアーティファクトであることがわかった.典型的なアーティファクトは,カルパイン活性化29–31)・小胞体ストレス32–34)・インフラマソーム35, 36)などの関与である(表1).これらのアーティファクトに対して行われていた創薬研究はむだであった可能性が高い.
4. App KIマウスの応用例—脳内Aβ代謝機構について—
I部2節で述べたように,ソマトスタチン受容体1および4がNEPの活性に関与する受容体であることを明らにした26).しかしながら,その下流のメカニズムについては明らかとなっておらず,その詳細が求められた.そこで,初代神経細胞共培養系モデルを構築し,ソマトスタチン添加後に網羅的なスクリーニングを行い,ENSA(α-endosulfine)がソマトスタチンシグナルの下流でNEPの活性を制御している因子であると同定した.次に,CRISPR/Cas9システムを利用し,ENSAのノックアウトマウスを作製したところ,マウス個体内においても,ENSAはNEPの活性を制御していることを明らかにした.ENSAの機能は,内因性のKATPチャネルのリガンドである.KATPチャネルはヘテロ八量体で存在し,スルホニルウレア受容体1, 2(SUR1およびSUR2AまたはB)と内向き整流カリウムチャネルKir6.1またはKir6.2の複合体である.次に,KATPチャネルのどのサブタイプがNEP活性制御に関わっているのかを調べるために,各受容体のノックアウトマウスを導入し解析を行ったところ,SUR1とKir6.2のノックアウトマウスにおいて,NEPの活性が強く下方調節されていることが明らかとなった.実は,この特定したKATPチャネルのサブタイプ(SUR1/Kir6.2)は膵β細胞に強く発現しており,インスリン分泌に関与している.ジアゾキシドはKATPチャネルを活性化することでインスリン分泌を抑制し,血糖上昇作用を示す薬剤であり,高インスリン血性低血糖症の治療薬として使用されている.この薬剤がNEPの活性上昇を介して,脳内に蓄積するAβ1–40およびAβ1–42の量を減らすのかどうか,AppNL–F KIラインを用いて,評価を行った.その結果,ジアゾキシドの長期的な投与で大脳皮質および海馬におけるAβの蓄積が減少し,障害された長期記憶が改善されていることを明らかにした.したがって,ジアゾキシドは血糖値コントロールに注意を払う必要があるが,ドラッグリポジショニングとして,有用なAD予防薬になる可能性が示された25).ジアゾキシドを従来の過剰発現によって作製されたADモデルマウスに投与すると,細胞内Aβの蓄積量は上昇するとの報告があったが37),ジアゾキシドで治療を施したAppNL–F KIラインでは,そのような現象は観察されなかった.
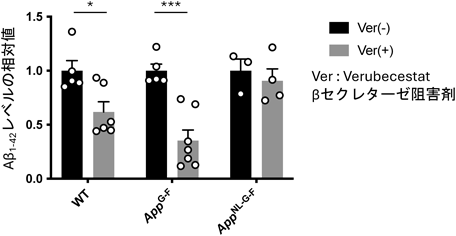
5. AppG–F KIおよびPsen1P117Lマウスの作製
これまでに作製してきたAppNL–F,AppNL–G–F KIマウスはいくつかの欠点がある.たとえば,スウェーデン変異(NL変異)は,βセクレターゼの切断部位近傍に存在し,総Aβ量を上昇させる効果がある.そのため,NL変異を含むApp KIマウスは,生理的なβセクレターゼの機能を解析するには不向きであり,AD予防薬としてβセクレターゼ阻害剤を正しく評価することが難しい.そこで,我々は,CRISPR/Cas9を用いてAppG–F KIラインの作製を試みた.その結果,AppG–F KIマウス脳内で月齢依存的なAβ1–42およびAβ3pE-42の沈着が確認された.Aβの蓄積速度はAppNL–G–Fマウスに比べ遅いが,βセクレターゼ阻害剤の効果を適切に評価することが可能である(図1)38).また,Aβ内部に存在する北極変異(G変異)はAβの構造を変化させ,より凝集を加速させる変異と考えられている.しかしながら,この変異があるAβはNEPによる分解抵抗性を示したことから39),Aβの凝集だけでなく,代謝機構にも影響を及ぼす可能性が示唆された.また,AppNL–FマウスのAβ42の蓄積は12か月齢より観察され始めるが,Aβプラークの密度を適切に評価するためには18か月齢ほど加齢させる必要がある.そのため,Aβ内部の変異に依存することなく,AppNL–Fマウスより速くAβの蓄積が観察されるマウスモデルが求められた.そこで,我々はADの家族性原因遺伝子として同定されたI部3節で記載したプレセニリン1(Psen1)に着目し,Psen1P117L KIマウスを創出した40).また,AppNL–Fマウスと交配することでAppNL–F;Psen1P117L/WTマウスを作製し,評価を行った.その結果,AppNL–G–Fマウスとほぼ同程度の速度で大脳皮質および海馬におけるAβ1–42の蓄積が観察された41).興味深いことに,AppNL–F;Psen1P117L/WTマウスは,より毒性が強いと考えられているAβ1–43の蓄積および,1-fluoro-2,5-bis(3-carboxy-4-hydroxystyryl)benzene(FSB)陽性のAβプラーク(βシート構造を持つ)の密度が高いことが明らかとなった42).これらの特徴は,AD患者脳においても観察されることから,よりAD病態に近いマウスモデルと考えられる.
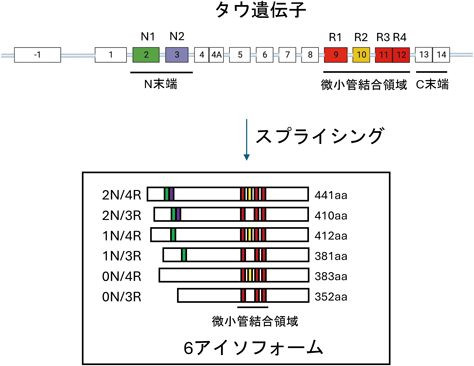
ADをはじめとするいくつかの神経変性疾患では,タウタンパク質の凝集体が神経細胞およびグリア細胞に蓄積する.これらの疾患をタウオパチーと称する.タウは微小管結合タンパク質として同定され,軸索に多く存在する.ヒト脳では,タウはN末端側のexon 2とexon 3,微小管結合領域に存在するexon 10の選択的スプライシングにより,六つのアイソフォームが発現している(図2).また,微小管結合領域が三つ(3-repeat:3R)と四つ(4-repeat:4R)のタウがほぼ同じ比率で発現し,認知症の疾患ごとに蓄積するタウの特徴が異なる.たとえば,ADや原発性加齢性タウオパチー(primary age-related tauopathy:PART)では3Rと4Rのタウが蓄積する.進行性核上性麻痺(progressive supranuclear palsy:PSP)や大脳皮質基底核変性症(corticobasal degeneration:CBD)では4Rタウ,ピック病(Pick disease:PiD)では3Rタウが蓄積することが剖検脳の解析より明らかになった.また,近年では,凝集したタウが細胞間を伝播する「伝播仮説」が提唱されており,タウの伝播能はin vitroおよびin vivoの解析によって,実験的なエビデンスが示されている43, 44).
疾患研究において,細胞および個体(実験動物)がどれほど,その患者に近い症状を呈するか否かが,その研究成果の解釈に直結する.そのため,モデル系の確立は各研究分野でも非常に重要な要素であり,疾患の発症メカニズム解明から創薬研究まで多岐にわたり使用される.ここからは,我々がこれまでに注力してきた,タウオパチーモデルマウスの開発から応用を概説する.
これまでのタウオパチー研究では,家族性前頭側頭葉変性症(FTLD)で同定された病原性変異を含むタウ遺伝子を過剰発現させたマウスモデルが,幅広く使用されてきた(表2).FTLDの中で,これまでに55以上の病原性変異がタウ遺伝子上で同定されており,異常タウが病態発症のトリガーとなっていることが考えられる.Godertらのグループは,Thy1.2プロモーター下にP301S変異を含んだ0N4Rタウを過剰発現させたTgマウスを作製した.このマウスは,3, 4か月齢よりタウの凝集体や大脳皮質の神経細胞死が検出され,記憶障害は2.5か月齢より観察される45).Leeらのグループも同様にP301S変異を組み込んだ1N4Rタウにプリオンプロモーターを用いて過剰発現させているTgマウスを作製した.このマウスは,6~8か月齢程度でタウの凝集体および脳萎縮が観察される46).一方Lewisらのグループは,CaMKIIプロモーター下流にtetracycline-controlled transactivator(tTA)を組み込んだマウスと,テトラサイクリン依存型転写調節因子下流にP301Lの変異を含む0N4Rのアイソフォームを組み込んだマウスを掛け合わせ,Tg(rTg4510)マウスを作製した.このマウスは,3~4か月齢にて大脳皮質の神経細胞内にタウの凝集体を認め,5.5か月齢より海馬CA1の神経細胞の減少が観察される.10か月齢より大脳皮質の萎縮が認められる.2.5~4か月齢の間で,空間記憶が障害されることがわかっている.興味深いことに,このTgマウスはDoxycycline(テトラサイクリン誘導体)による遺伝子発現の調節が可能であり,Doxycycline投与後,タウの遺伝子発現低下に伴い,凝集性タウの沈着量は増え続けるが,認知機能が改善されることが報告された47).これらの傍証は,凝集性タウの沈着は神経細胞毒性が少ないことを示し,タウオリゴマー毒性を強く支持していると考えられる.また,Peter, Duffらのグループはヒトタウ遺伝子を過剰発現したTg(hTau)マウスを作製した.このマウスは,加齢依存的なリン酸化タウの蓄積,および記憶障害を示した48).最近では,東京都医学総合研究所の長谷川チームは,CRISPR/Cas9を用いて,3Rと4Rマウスタウが同等のレベルで発現するマウスを作製した.ADやCBD, PiD患者脳から界面活性剤不溶性画分を抽出し,それぞれのタウ線維(鋳型となるシードを含む)を作製したマウスへ脳内に注入したところ,アイソフォーム特異的なシード依存性伝播が認められた49).
表2 これまでのタウオパチーモデルマウス| マウス | 遺伝的背景 | プロモーター | 変異 | タウ凝集体蓄積 | 神経細胞消失 | グリオーシス | 認知機能障害 |
|---|
| JNPL3 | C57B/6×DBA/2×SW | Prion | P301L | 4.5か月齢 | 10~14か月齢 | 10か月齢 | 不明 |
| hTau.P301S | CBAxC57BL/6 | Thy1.2 | P301S | 3~4か月齢 | 3か月齢 | 5~6か月齢 | 2.5か月齢 |
| PS19 | C57B/6×C3H | Prion | P301S | 6か月齢 | 9~12か月齢 | 6か月齢 | 6~7か月齢 |
| rTg4510 | 129S6×FVB | CamKII | P301L | 4か月齢 | 5.5か月齢 | 不明 | 3か月齢 |
| 3xTg | C57BL/6;129X1/SvJ;129S1/Sv | Thy1.2 | P301L | 12か月齢 | 不明 | 7か月齢 | 4か月齢 |
| hTau | C57BL/6 | MAPT | — | 9か月齢 | 10~14か月齢 | 不明 | 6か月齢 |
野生型マウスは4Rタウのみ発現しており,ヒト脳におけるタウの発現パターンと異なるため,ヒト病理を再現するには不向きである.そこで,我々は相同的組換えを用いてマウスタウ遺伝子をヒトタウに置換した,ヒトタウノックイン(MAPT KI)マウスを作製した.このマウスは六つのアイソフォームのタウが発現し,その局在はマウスタウと同等であることが確かめられた.また,認知機能や不安様行動は野生型マウスとほぼ同等であることから,タウ遺伝子のヒト化は脳高次機能に影響を与えないことが示唆された50–52).次に,ADの主原因であるAβとヒト化タウとの関連を調べるために,我々の研究室で作製したApp KIマウス31)とMAPT KIマウスを交配し(double knock-in:dKI),解析を行った.その結果,dKIマウスにおいて,タウのリン酸化レベルがMAPT KIマウスに比べて,上昇していることを明らかにした.興味深いことに,AD患者脳から得られたタウ線維を野生型マウスとMAPT KIマウスへ播種した場合,MAPT KIマウスの方が,より広範囲にタウ病理(AT8陽性のシグナル)が拡散していることが明らかとなった.このことから,MAPT KIマウスはよりヒトに近い状態で,タウ病理の広がりを解析できるモデルであることが実証された.また,タウ病理の広がりは,アミロイド病理によって加速することも明らかとなり,dKIマウスは「アミロイド病理」と「タウ病理」の分子関連を調べるツールとして,有用であることが示された43).しかしながら,dKIマウスは,AD患者脳の病理所見である神経原線維変化や神経細胞死は観察されなかった.これらの変化はヒト脳内で10年以上を経て生じるので,何らかの工夫をしなければマウスの寿命期間内に再現することは難しいのであろう.
タウをヒト化したマウスは,盛んに開発が展開されており,アメリカのNIA(National institute on Aging)が中心となり進めているThe Model AD Consortiumにおいて,Koobらの研究室はプロモーター領域を含むマウスタウ遺伝子をヒトタウ遺伝子と置換した,タウノックインマウスの作製に成功している.また,Cynisらの研究室では,マウスタウのexon 2からexon 10に相応する部分をヒト化したノックインマウスを作製した.両者のノックインマウスも3Rおよび4Rタウの発現が認められ,前臨床AD研究に活用されている53).
新しいタウオパチーモデルマウスを作製するために,我々がこれまでに作製したMAPT KIマウスにFTLD-17で同定された病原性変異の導入を試みた.方法としては,CRISPR/Cas9の改変型であり,より容易に点変異導入を可能にした“塩基編集技術”(base editor:BE)を利用した.この技術はハーバード大学のLiuらによって作製された54).BEは,核酸の代謝に関連するデアミナーゼ(脱アミノ化酵素)の一部を変異Cas9と連結させることで,DNAの二本鎖切断を起こさずに,塩基を変換することができる.ガイドRNAの標的配列に対して,どの部分の塩基をどのように編集できるのかは,これまで開発されてきた塩基編集技術によって異なるため,実験の目的に合致した適切な酵素を使用する必要がある.たとえば,シトシンBEはシトシン(C)をチミン(T)に変換することが可能であり,アデニンBEはアデニン(A)をグアニン(G)へと変換することができる.
次に,ガイドRNA(single guide RNA:sgRNA)の探索を行った.P301およびtron10+3(G>A)の変異部位に相応するsgRNAを設計することに成功し,MAPT KIマウスから得られた受精卵にBEツールと設計したsgRNAを注入した.その結果,単一および複数種類の病原性変異がMAPT遺伝子上に導入された変異MAPT KIマウスを獲得した(表3).本稿では,解析が進んでいるMAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスの表現型を解説する.
表3 これまでに著者らが作製したタウオパチーモデルマウス| マウス | 遺伝的背景 | プロモーター | 部位 | 種類 | コーディングor
ノンコーディング | コドン | 期待される変異の効果 |
|---|
| P301L | C57B/6 | Mapt | exon 10 | Substitution | コーディング | CCG(P) to TTG(L) | 微小管重合の抑制
タウの凝集促進 |
| P301S | C57B/6 | Mapt | exon 10 | Substitution | コーディング | CCG(P) to TCG(S) | 微小管重合の抑制
タウの凝集促進 |
| Intron10+3G>A | C57B/6 | Mapt | intron 10 | Substitution | ノンコーディング | G to A | 4Rタウの上昇 |
| P301L;Intron10+3G>A | C57B/6 | Mapt | exon 10+intron 10 | Substitution | コーディング+
ノンコーディング | CCG(P) to TTG(L)/G to A | 微小管重合の抑制
タウの凝集促進
4Rタウの上昇 |
| P301S;Intron10+3G>A | C57B/6 | Mapt | exon 10+intron 10 | Substitution | コーディング+
ノンコーディング | CCG(P) to TCG(S)/G to A | 微小管重合の抑制
タウの凝集促進
4Rタウの上昇 |
| S305N;Intron10+3G>A | C57B/6 | Mapt | exon 10+intron 10 | Substitution | コーディング+
ノンコーディング | AGT(S) to AAT(N)/G to A | 4Rタウの上昇 |
10. MAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスの組織・生化学的解析
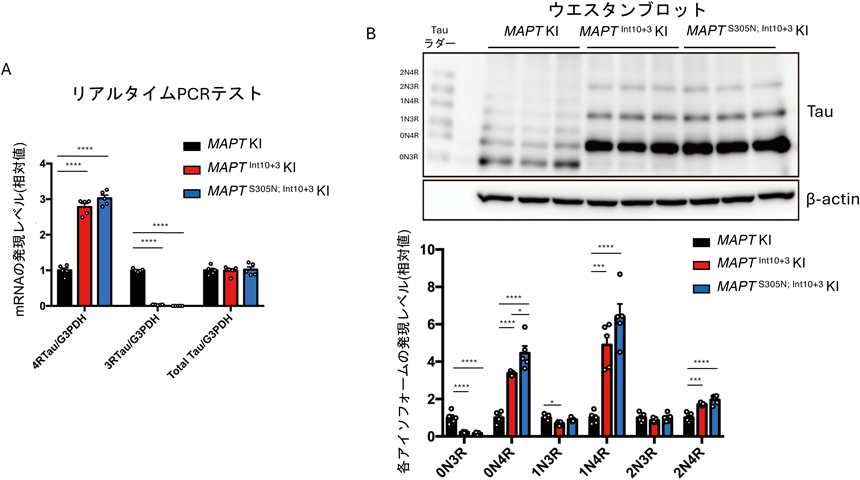
これまでの症例報告では,Intron10+3(G>A)変異やS305N変異を持つ患者は,人格の変化や認識・記憶障害,方向感覚の喪失,運動障害などさまざまな症状が報告されている.病理学的な特徴としては,前頭葉および側頭葉の神経細胞内およびグリア細胞内にタウの凝集が強くみられ,脳萎縮を伴う.また,病原性変異の効果としては,exon 10のスプライシングに影響を与え,3R/4Rタウの比率を変化させることが細胞を用いた実験で明らかにされている55).したがって,作製したMAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスにおいて3R/4Rタウの発現比率をリアルタイムPCRやウエスタンブロット(WB)を用いて調べた.その結果,作製したマウスにおいて4Rタウの発現がコントロールのマウスに比べ上昇し,3Rタウの発現が低下していた(図3).
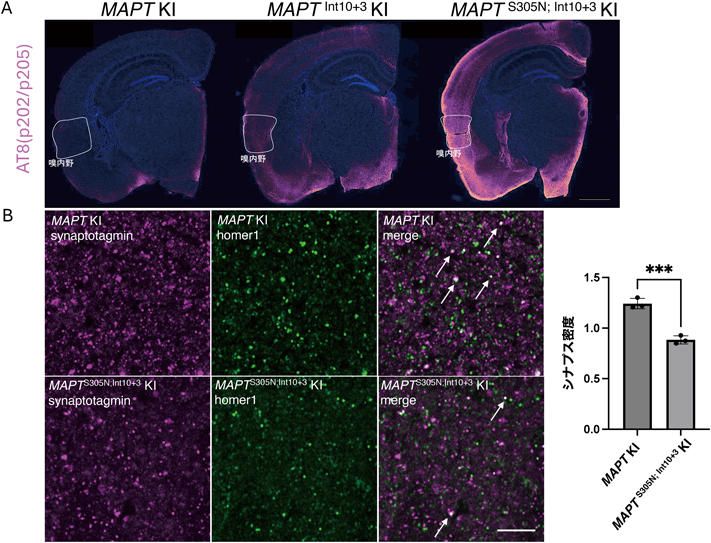
次に,病理学的特徴の一つであるリン酸化を調べるために,WBやリン酸化プロテオーム解析を行った結果,MAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスはタウのリン酸化レベルが有意に上昇していることを明らかにした.リン酸化タウ特異的抗体(AT8)を用いた免疫染色では,嗅内野の神経細胞において,特に強い陽性シグナルが観察された(図4A).興味深い点は,これらマウスの特性として,凝集したタウは観察されず,タウのシード活性がない点である.オリゴマータウ特異的抗体(TOC1, T22)を用いた免疫染色では,陽性のシグナルは観察されず,サルコシル不溶性画分においてもタウは検出されなかった.また,テキサス大学Southwestern Medical centerのDiamond研究室によって作製されたバイオセンサー細胞を用いた実験においても,MAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスの脳試料からシード活性があるタウを検出することができなかった56).
次に,神経細胞毒性の評価として,嗅内野におけるシナプスの数を分析した.これは,前シナプスマーカー(Synaptotagmin),後シナプスマーカー(Homer1)の二重染色を行い,共局在しているシグナルの密度を解析した.その結果,MAPTS305N; Int10+3 KIマウスの嗅内野において,顕著にシナプスの数が減少していることが明らかとなった(図4B).また,神経変性を検出するための,アミノ酸-銅-銀染色を用いたところ,MAPTS305N; Int10+3 KIは嗅内野において顕著に陽性シグナルが増えていることが確認された.
11. MAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスの行動学的解析
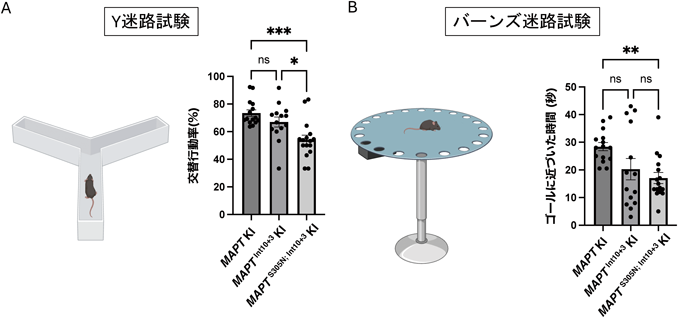
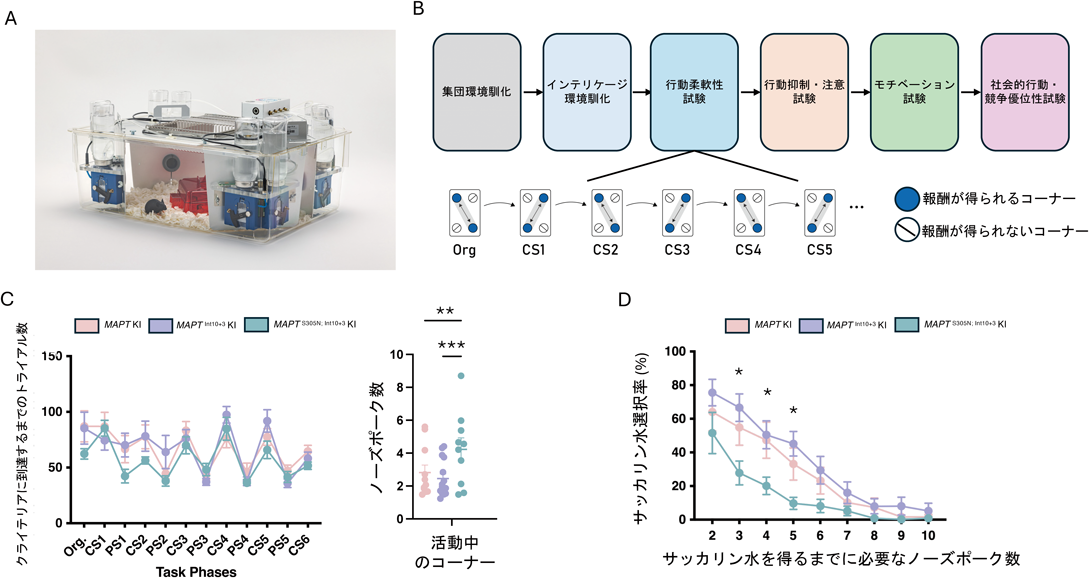
続いて,作製したMAPTInt10+3 KIとMAPTS305N; Int10+3 KIマウスの行動学的特徴を調べた.本実験では,Y迷路試験,オープンフィールド,新規物体認識および位置認識試験,バーンズ迷路試験,ロタロット試験を行い,認識・記憶や運動機能の評価を行った.その結果,Y迷路試験やバーンズ迷路試験において,MAPTS305N; Int10+3 KIマウスの短期記憶(ワーキングメモリー)や長期記憶の障害が観察された(図5).さらに深く行動学的特徴を調べるために,インテリケージ解析を行った.インテリケージシステムは,全自動・集団飼育環境下マウス行動表現型解析装置であり,動物への刺激,実験者への負担,実験者の違いによる結果のばらつきなどを軽減することができる(図6A)57).また,飼育環境と試験環境が同一のため,十分に慣れた日常的環境化での長期試験が可能である.同一プラットフォームで,多様な認知機能試験を評価することが可能であり,本実験では,行動柔軟性(Complete/Partial Shifts Shuffled test:SP-FLEX)や行動抑制,注意,モチベーションに関する試験を行った(図6B).SP-FLEXにおいて,マウスは報酬を効率よく得るために,四つのコーナーを特定のルールに従って訪れる必要がある.たとえば,対角線上に位置する二つのコーナーを交互に行き来することで効率的に報酬を得られることを学習する.個々の成績はSPRTと呼ばれる統計学的検討法を用いて,正答率を算出する.ある一定の値を超えた場合,マウスは自動的に次のルールに従う必要がある.SP-FLEXでは,マウスがこの状況にどれほど柔軟に対応できるのかという高度な認知能力(行動柔軟性)を評価することができる(図6B).また,試験期間中どれくらいコーナーに訪れたか,どれほど活動中のコーナーで報酬を得られないノーズポーク(水を得るためにマウスが穴に鼻を近づける動作)をしたかを計測することで,常同行動を解析することも可能である.MAPTS305N; Int10+3 KIマウスの行動柔軟性はコントロールマウスと同等であったが,意味のないノーズポーク数が有意に上昇していることから,常同行動が増えていることが明らかとなった(図6C).また,サッカリン水を用いたモチベーションテスト(effort-based choice test)では,MAPTS305N; Int10+3 KIマウスはコントロールマウスに比べ,報酬を得る意欲が低いことがわかった(図6D).これらの表現型はFTLDの症状と部分的に一致しており,FTLDモデルマウスとしての有用性を支持する結果となった.
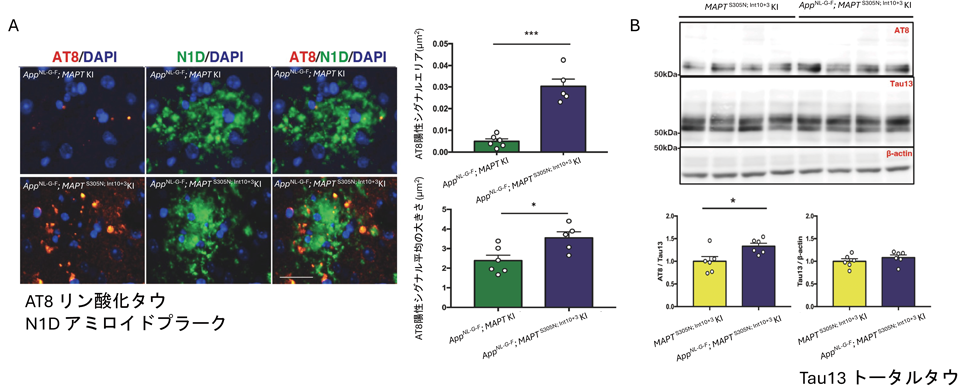
次に,MAPTS305N; Int10+3 KIマウスとI部3節にて紹介したAppNL–G–F KIマウスを交配し,アミロイド病理とタウ病理間の相互作用を調べた.ADの病理学的特徴の一つとして,神経変性突起(dystrophic neurites:DN)と呼ばれるものが,アミロイドプラーク周辺に観察される.DNには,リン酸化タウの蓄積が観察される.興味深いことに,AppNL–G–F;MAPTS305N; Int10+3 KIマウスは,DNへのリン酸化タウの蓄積がAppNL–G–F;MAPT KIマウスに比べ,増えていることが明らかとなった(図7A).また,WBにおいてもタウのリン酸化が上昇していることが明らかとなり,Aβ1–42の蓄積は,タウのリン酸化とリンクしている可能性が示唆された(図7B).しかしながら,Aβがどのようにタウのリン酸化を上昇させているのか,分子メカニズムを解明するには,今後,詳細なアプローチが必要である.
認知症を伴う神経変疾患の病理形成機構はいまだに不明な点が多く,根本的な治療法の確立も実現できていないのが現状である.これまでに,過剰発現でタウ病理を再現するマウスモデルは,タウオパチー研究を着実に発展させてきたが,過剰発現による個体間のタウ病理のばらつきや寿命が短いこと,内在性遺伝子の破壊による表現型への影響などさまざまなアーティファクトの存在が示唆されている.そのため,より適切なモデルを用いて,病理へのアプローチを再考することも必要である.特に,タウオパチーの中でもタウのリン酸化を含む翻訳後修飾や凝集,細胞間伝播能など,どのようなタウがどのように毒性を発揮するのか,どのように神経細胞死を導くのかについては,いまだ議論されている.55以上同定されてきた,MAPT遺伝子上の病原性変異はどのように病態を形成するのか,それぞれ適切なモデルを作製し,評価する必要がある.また,ADの根本的治療法創出のために,アミロイド病理とタウ病理の相互作用を詳細に解析することも重要である.本研究で作製したモデルが,少しでも病態への理解を深め,治療法の開発に貢献することを期待する.
理研の神経老化研究チームは今のところ,2025年3月をもっていったん終了する予定である.その後は,言論の自由が守られる場所であれば世界の何処にでも行って,下記のチームからなる「前臨床性アルツハイマー病研究部門」を設立し,安価で効果的かつ副作用のない創薬に挑み「アルツハイマー病のない世界」を創りたい12).ただし,必要なツールはすべて理研にそろっており,数兆円を超える知財を生むと期待されるので,できれば日本で実行したい.
- 1) 先制医療研究チーム:我々が取得した特許「ソマトスタチン受容体(特許第7099717号)」に基づきGタンパク質共役型受容体創薬を目指す.また,ソマトスタチンの作用はKATPチャンネルを介するので,脳特異的KATPチャンネルを標的とした創薬も行う.
- 2) タウ病理研究チーム:新規動物モデルのES/iPSC由来の神経細胞を用い,抗タウ病理化合物のin vitroスクリーニングも行い,in vivo系で検証する.さらに,ユビキチン-プロテアソームやオートファジーによる選択的タンパク質分解技術(Proteolysis-Targeting Chimera:PROTAC)を用いて病理学的タウを排除する創薬を実行する58–64).
- 3) オミックス研究チーム:近年,一細胞単位で,あるいは,局所空間で個々の遺伝子発現を定量化する新しい手法が開発された(Single cell RNA sequencing, Spatial transcriptomics等).Aβがタウ病理を加速するモデルを作製することに成功した12)ので(図7),Aβ-タウaxisに関与する因子を同定し第三の創薬標的とする.また,アルツハイマー病の発症に寄与する多くの遺伝子(約70個)が同定され,これらの遺伝子多型を数学的に処理することによって約80%の確率で発症年齢を予測することが可能になるとの報告がある.ただし,これは英国における仮説なので,日本人に関して,再度,検証する必要がある.大規模なゲノム解析データやアルツハイマー病患者,各種動物モデルから採取した脳試料・体液試料を詳細に解析することで,アルツハイマー病の発症を正確に予測する.これにより,上記の「第一の戦略(先制医療)」や「第二の戦略(タウ病理抑制)」や「第三の戦略(Aβtau axis抑制)」をもって,個々のリスクに応じ適切に治療介入できるパラダイムを構築する.
- 4) 霊長類モデル研究チーム:我々は,最近,実験動物中央研究所,広島大学との共同研究で,世界初の非ヒト霊長類アルツハイマー病モデルとして,遺伝子改変マーモセットの作製に成功した.マーモセットは遺伝学的背景,脳の構造や機能,社会的行動,認知行動,内分泌代謝機能,免疫機能など,多くの面でヒトに近く,今後のアルツハイマー病研究において中心的な役割を担うことが期待される.アルツハイマー病マーモセットモデルの前臨床段階に着目し,未知の表現型の検索,超早期の神経回路の障害の検出を目指し,上記の他研究チームと連携することで新たなバイオマーカー・治療戦略の確立を目指す.さらに,製薬や臨床の現場と連携し,準臨床試験を加速する.ヒトを対象とした前臨床性アルツハイマー病治療薬の臨床試験は約5~10年を要するといわれるが,非ヒト霊長類モデルを用いれば,これが1~2年に短縮されるだろう.また,各国の研究者と連携し,積極的に共同研究を展開することにより,世界のアルツハイマー病研究の拠点となることを目指す.
振り返ると,我々の研究は多くの方々との出会いによって始まり,展開してきた.これらの出会いがなければ,私どもの研究はここまで進まなかったであろう.綿村と西道の出会いも邂逅だったと思っている.綿村が西道研に入ったころの印象は,緻密な研究計画を立案し,慎重に実行し,正確なロジックを持って結論を導き出すとのできる優秀な研究者であった.一方で,彼の唯一最大の弱点は英語力であった.そこで,英語圏への留学を強く勧めた.その成果は,現在投稿中の論文原稿執筆や理研CBSにおけるforumで発揮されているようである.科学研究者である限り,英語力は必須である.国際学会における発表・講演・質疑応答だけでなく,座長としてシンポジウム等を仕切る能力が求められる.綿村が将来PIとなったときに今回の留学経験は大いに生きるであろう.
最後に,何よりもこれまで出会ってきたすべての方々(私のメンタルを鍛えてくれたストレッサーを含めて)に御礼申し上げたい.また,その多くが鬼籍に入られたことに対して暗澹とした気持ちになることはあるが,それはすべての人間の運命であり,いずれ自分にも訪れることである.また,人類に害をなす者たちもいずれ老いて去って行くことはむしろ吉報であろう.私自身は,今後どのような生き様と死に様を示すにしても,残る方々に恥じることのない道を歩みたい.
引用文献References
1) Miyawaki, A., Llopis, J., Heim, R., McCaffery, J.M., Adams, J.A., Ikura, M., & Tsien, R.Y. (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature, 388, 882–887.
2) Denk, W., Strickler, J.H., & Webb, W.W. (1990) Two-photon laser scanning fluorescence microscopy. Science, 248, 73–76.
3) Helmchen, F. & Denk, W. (2005) Deep tissue two-photon microscopy. Nat. Methods, 2, 932–940.
4) Saido, T.C., Nagao, S., Shiramine, M., Tsukaguchi, M., Sorimachi, H., Murofushi, H., Tsuchiya, T., Ito, H., & Suzuki, K. (1992) Autolytic transition of mu-calpain upon activation as resolved by antibodies distinguishing between the pre- and post-autolysis forms. J. Biochem., 111, 81–86.
5) Saido, T.C., Yokota, M., Nagao, S., Yamaura, I., Tani, E., Tsuchiya, T., Suzuki, K., & Kawashima, S. (1993) Spatial resolution of fodrin proteolysis in postischemic brain. J. Biol. Chem., 268, 25239–25243.
6) Saido, T.C., Sorimachi, H., & Suzuki, K. (1994) Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J., 8, 814–822.
7) Saido, T.C., Yokota, M., Maruyama, K., Yamao-Harigaya, W., Tani, E., Ihara, Y., & Kawashima, S. (1994) Spatial resolution of the primary beta-amyloidogenic process induced in postischemic hippocampus. J. Biol. Chem., 269, 15253–15257.
8) Saido, T.C., Yokota, M., Maruyama, K., Yamaoharigaya, W., Tani, E., Ihara, Y., & Kawashima, S. (1994) Spatial-resolution of the primary beta-amyloidogenic process-induced in postischemic hippocampus. J. Biol. Chem., 269, 15253–15257.
9) Mori, H., Takio, K., Ogawara, M., & Selkoe, D.J. (1992) Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J. Biol. Chem., 267, 17082–17086.
10) Saido, T.C., Iwatsubo, T., Mann, D.M., Shimada, H., Ihara, Y., & Kawashima, S. (1995) Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3pE), in senile plaques. Neuron, 14, 457–466.
11) Saido, T.C., Yamao-Harigaya, W., Iwatsubo, T., & Kawashima, S. (1996) Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci. Lett., 215, 173–176.
12) Saido, T.C. (2024) Alzheimer’s Disease Research Guide: Animal Models for Understanding Mechanisms and Medications, Elsevier Inc., Cambridge, MA.
13) DeMattos, R.B., Bales, K.R., Cummins, D.J., Dodart, J.C., Paul, S.M., & Holtzman, D.M. (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA, 98, 8850–8855.
14) Sims, J.R., Zimmer, J.A., Evans, C.D., Lu, M., Ardayfio, P., Sparks, J., Wessels, A.M., Shcherbinin, S., Wang, H., Monkul Nery, E.S., et al.; TRAILBLAZER-ALZ 2 Investigators. (2023) Donanemab in early symptomatic Alzheimer disease: The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA, 330, 512–527.
15) Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., Kawashima-Morishima, M., Lee, H.J., Hama, E., Sekine-Aizawa, Y., et al. (2000) Identification of the major Abeta1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med., 6, 143–150.
16) Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N.P., Gerard, C., Hama, E., Lee, H.J., & Saido, T.C. (2001) Metabolic regulation of brain Abeta by neprilysin. Science, 292, 1550–1552.
17) Iwata, N., Tsubuki, S., Takamura, R., Watamura, N., Kakiya, N., Fujioka, R., Mihira, N., Sekiguchi, M., Watanabe-Iwata, K., Kamano, N., et al. (2024) Metabolic resistance of Aβ3pE-42, target epitope of the anti-Alzheimer therapeutic antibody, donanemab. bioRxiv 2024.01.30.578111.
18) Qiu, W.Q., Walsh, D.M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M.B., Rosner, M.R., Safavi, A., Hersh, L.B., & Selkoe, D.J. (1998) Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem., 273, 32730–32738.
19) Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E.A., Frosch, M.P., Eckman, C.B., Tanzi, R.E., Selkoe, D.J., & Guenette, S. (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA, 100, 4162–4167.
20) Leissring, M.A., Farris, W., Chang, A.Y., Walsh, D.M., Wu, X., Sun, X., Frosch, M.P., & Selkoe, D.J. (2003) Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron, 40, 1087–1093.
21) Wightman, D.P., Jansen, I.E., Savage, J.E., Shadrin, A.A., Bahrami, S., Holland, D., Rongve, A., Borte, S., Winsvold, B.S., Drange, O.K., et al.; 23andMe Research Team. (2021) A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet., 53, 1276–1282.
22) Saito, T., Iwata, N., Tsubuki, S., Takaki, Y., Takano, J., Huang, S.M., Suemoto, T., Higuchi, M., & Saido, T.C. (2005) Somatostatin regulates brain amyloid beta peptide A beta42) through modulation of proteolytic degradation. Nat. Med., 11, 434–439.
23) Davies, P., Katzman, R., & Terry, R.D. (1980) Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature, 288, 279–280.
24) Dawbarn, D., Rossor, M.N., Mountjoy, C.Q., Roth, M., & Emson, P.C. (1986) Decreased somatostatin immunoreactivity but not neuropeptide Y immunoreactivity in cerebral cortex in senile dementia of Alzheimer type. Neurosci. Lett., 70, 154–159.
25) Watamura, N., Kakiya, N., Nilsson, P., Tsubuki, S., Kamano, N., Takahashi, M., Hashimoto, S., Sasaguri, H., Saito, T., & Saido, T.C. (2022) Somatostatin-evoked Aβ catabolism in the brain: Mechanistic involvement of α-endosulfine-K (ATP) channel pathway. Mol. Psychiatry, 27, 1816–1828.
26) Nilsson, P., Sörgjerd, K., Kakiya, N., Sasaguri, H., Watamura, N., Shimozawa, M., Tsubuki, S., Zhou, Z., Loera-Valencia, R., Takamura, R., et al. (2020) Somatostatin receptor subtypes 1 and 4 redundantly regulate neprilysin, the major amyloid-beta degrading enzyme in brain. bioRxiv. 2020.05.09.085795.
27) Lichtenthaler, S.F., Wang, R., Grimm, H., Uljon, S.N., Masters, C.L., & Beyreuther, K. (1999) Mechanism of the cleavage specificity of Alzheimer’s disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA, 96, 3053–3058.
28) Guerreiro, R.J., Baquero, M., Blesa, R., Boada, M., Bras, J.M., Bullido, M.J., Calado, A., Crook, R., Ferreira, C., Frank, A., et al. (2010) Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging, 31, 725–731.
29) Higuchi, M., Iwata, N., Matsuba, Y., Takano, J., Suemoto, T., Maeda, J., Ji, B., Ono, M., Staufenbiel, M., Suhara, T., et al. (2012) Mechanistic involvement of the calpain-calpastatin system in Alzheimer neuropathology. FASEB J., 26, 1204–1217.
30) Saito, T., Matsuba, Y., Yamazaki, N., Hashimoto, S., & Saido, T.C. (2016) Calpain activation in Alzheimer’s model mice is an artifact of APP and presenilin overexpression. J. Neurosci., 36, 9933–9936.
31) Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., Iwata, N., & Saido, T.C. (2014) Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci., 17, 661–663.
32) Hashimoto, S., Ishii, A., Kamano, N., Watamura, N., Saito, T., Ohshima, T., Yokosuka, M., & Saido, T.C. (2018) Endoplasmic reticulum stress responses in mouse models of Alzheimer’s disease: Overexpression paradigm versus knockin paradigm. J. Biol. Chem., 293, 3118–3125.
33) Hashimoto, S. & Saido, T.C. (2018) Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol., 8, 8.
34) Sadleir, K.R., Popovic, J., & Vassar, R. (2018) ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem., 293, 18434–18443.
35) Heneka, M.T., Kummer, M.P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., Griep, A., Axt, D., Remus, A., Tzeng, T.C., et al. (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature, 493, 674–678.
36) Srinivasan, S., Kancheva, D., De Ren, S., Saito, T., Jans, M., Boone, F., Vandendriessche, C., Paesmans, I., Maurin, H., Vandenbroucke, R.E., et al. (2024) Inflammasome signaling is dispensable for ß-amyloid-induced neuropathology in preclinical models of Alzheimer’s disease. Front. Immunol., 15, 1323409.
37) Liu, D., Pitta, M., Lee, J.H., Ray, B., Lahiri, D.K., Furukawa, K., Mughal, M., Jiang, H., Villarreal, J., Cutler, R.G., et al. (2010) The KATP channel activator diazoxide ameliorates amyloid-β and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer’s disease. J. Alzheimers Dis., 22, 443–457.
38) Watamura, N., Sato, K., Shiihashi, G., Iwasaki, A., Kamano, N., Takahashi, M., Sekiguchi, M., Mihira, N., Fujioka, R., Nagata, K., et al. (2022) An isogenic panel of App knock-in mouse models: Profiling β-secretase inhibition and endosomal abnormalities. Sci. Adv., 8, eabm6155.
39) Tsubuki, S., Takaki, Y., & Saido, T.C. (2003) Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet, 361, 1957–1958.
40) Sasaguri, H., Nagata, K., Sekiguchi, M., Fujioka, R., Matsuba, Y., Hashimoto, S., Sato, K., Kurup, D., Yokota, T., & Saido, T.C. (2018) Introduction of pathogenic mutations into the mouse Psen1 gene by Base Editor and Target-AID. Nat. Commun., 9, 2892.
41) Sato, K., Watamura, N., Fujioka, R., Mihira, N., Sekiguchi, M., Nagata, K., Ohshima, T., Saito, T., Saido, T.C., & Sasaguri, H. (2021) A third-generation mouse model of Alzheimer’s disease shows early and increased cored plaque pathology composed of wild-type human amyloid β peptide. J. Biol. Chem., 297, 101004.
42) Saito, T., Suemoto, T., Brouwers, N., Sleegers, K., Funamoto, S., Mihira, N., Matsuba, Y., Yamada, K., Nilsson, P., Takano, J., et al. (2011) Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci., 14, 1023–1032.
43) Saito, T., Mihira, N., Matsuba, Y., Sasaguri, H., Hashimoto, S., Narasimhan, S., Zhang, B., Murayama, S., Higuchi, M., Lee, V.M.Y., et al. (2019) Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem., 294, 12754–12765.
44) Takeda, S., Wegmann, S., Cho, H., DeVos, S.L., Commins, C., Roe, A.D., Nicholls, S.B., Carlson, G.A., Pitstick, R., Nobuhara, C.K., et al. (2015) Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun., 6, 8490.
45) Allen, B., Ingram, E., Takao, M., Smith, M.J., Jakes, R., Virdee, K., Yoshida, H., Holzer, M., Craxton, M., Emson, P.C., et al. (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci., 22, 9340–9351.
46) Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S.M., Iwata, N., Saido, T.C., Maeda, J., Suhara, T., Trojanowski, J.Q., & Lee, V.M. (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron, 53, 337–351.
47) Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., DeTure, M., Ramsden, M., McGowan, E., et al. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science, 309, 476–481.
48) Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K.L., Barde, Y.A., Duff, K., & Davies, P. (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem., 86, 582–590.
49) Hosokawa, M., Masuda-Suzukake, M., Shitara, H., Shimozawa, A., Suzuki, G., Kondo, H., Nonaka, T., Campbell, W., Arai, T., & Hasegawa, M. (2022) Development of a novel tau propagation mouse model endogenously expressing 3 and 4 repeat tau isoforms. Brain, 145, 349–361.
50) Hashimoto, S., Matsuba, Y., Kamano, N., Mihira, N., Sahara, N., Takano, J., Muramatsu, S.I., Saido, T.C., & Saito, T. (2019) Tau binding protein CAPON induces tau aggregation and neurodegeneration. Nat. Commun., 10, 2394.
51) Watamura, N., Sato, K., & Saido, T.C. (2022) Mouse models of Alzheimer’s disease for preclinical research. Neurochem. Int., 158, 105361.
52) Benskey, M.J., Panoushek, S., Saito, T., Saido, T.C., Grabinski, T., & Kanaan, N.M. (2023) Behavioral and neuropathological characterization over the adult lifespan of the human tau knock-in mouse. Front. Aging Neurosci., 15, 1265151.
53) Barendrecht, S., Schreurs, A., Geissler, S., Sabanov, V., Ilse, V., Rieckmann, V., Eichentopf, R., Künemund, A., Hietel, B., Wussow, S., et al. (2023) A novel human tau knock-in mouse model reveals interaction of Abeta and human tau under progressing cerebral amyloidosis in 5xFAD mice. Alzheimers Res. Ther., 15, 16.
54) Komor, A.C., Kim, Y.B., Packer, M.S., Zuris, J.A., & Liu, D.R. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 533, 420–424.
55) Hasegawa, M., Smith, M.J., Iijima, M., Tabira, T., & Goedert, M. (1999) FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett., 443, 93–96.
56) Holmes, B.B., Furman, J.L., Mahan, T.E., Yamasaki, T.R., Mirbaha, H., Eades, W.C., Belaygorod, L., Cairns, N.J., Holtzman, D.M., & Diamond, M.I. (2014) Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA, 111, E4376–E4385.
57) Lipp, H.P., Krackow, S., Turkes, E., Benner, S., Endo, T., & Russig, H. (2023) IntelliCage: the development and perspectives of a mouse- and user-friendly automated behavioral test system. Front. Behav. Neurosci., 17, 1270538.
58) Guardigni, M., Pruccoli, L., Santini, A., Simone, A., Bersani, M., Spyrakis, F., Frabetti, F., Uliassi, E., Andrisano, V., Pagliarani, B., et al. (2023) PROTAC-induced glycogen synthase kinase 3β degradation as a potential therapeutic strategy for Alzheimer’s disease. ACS Chem. Neurosci., 14, 1963–1970.
59) Inuzuka, H., Liu, J., Wei, W., & Rezaeian, A.H. (2022) PROTACs technology for treatment of Alzheimer’s disease: Advances and perspectives. Acta Materia Med., 1, 24–41.
60) Jangampalli Adi, P. & Reddy, P.H. (2021) Phosphorylated tau targeted small-molecule PROTACs for the treatment of Alzheimer’s disease and tauopathies. Biochim. Biophys. Acta Mol. Basis Dis., 1867, 166162.
61) Kargbo, R.B. (2019) Treatment of Alzheimer’s by PROTAC-Tau protein degradation. ACS Med. Chem. Lett., 10, 699–700.
62) Ma, K., Han, X.X., Yang, X.M., & Zhou, S.L. (2021) Proteolysis targeting chimera technology: A novel strategy for treating diseases of the central nervous system. Neural Regen. Res., 16, 1944–1949.
63) Tashima, T. (2023) Proteolysis-Targeting Chimera (PROTAC) delivery into the brain across the blood-brain barrier. Antibodies (Basel), 12, 43.
64) Wang, W., Zhou, Q., Jiang, T., Li, S., Ye, J., Zheng, J., Wang, X., Liu, Y., Deng, M., Ke, D., et al. (2021) A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics, 11, 5279–5295.
65) Watamura, N., Foiani, M., Bez, S., Bourdenx, M., Frodsham, C., Camporesi, E., Brinkmalm, G., Zetterberg, H., Patel, S., Kamano, N., et al. (2024) In vivo hyperphosphorylation of tau is associated with synaptic loss and behavioral abnormalities in the absence of tau seeds. Nat. Neurosci., in press.
著者紹介Author Profile
 綿村 直人(わたむら なおと)
綿村 直人(わたむら なおと)University College London Dementia Research Institute Research Fellow. 理学博士.
略歴2018年早稲田大学大学院先進理工学研究科生命医科学専攻修了,博士課程より理化学研究所脳神経科学研究センター神経老化制御研究チーム(西道ラボ)にジュニアリサーチアソシエイトとして所属する.博士取得後は,研究員として西道ラボに所属し,2022年よりUniversity College LondonのDementia Research Institute(Duff, Karen Lab)にResearch Fellowとして所属する.
研究テーマと抱負基礎研究から認知症の病態機序を紐解くことを目指し,研究を進めている.特に,脳内アミロイドベータ代謝機構の解明とモデル動物の開発に力を入れている.
趣味旅行,サッカー,パブ巡り.
 西道 隆臣(さいどう たかおみ)
西道 隆臣(さいどう たかおみ)理化学研究所脳神経科学センター チームリーダー.薬学博士.
略歴1982年筑波大学生物学類卒業,88年東京大学大学院薬学系研究科博士課程修了,88~97年東京都臨床医学総合研究所遺伝情報研究部門主事,97年~現在理化学研究所脳神経科学センター(脳科学総合研究センター)チームリーダー.早稲田大学理工学部客員教授,慶應義塾大学医学部客員教授,日本認知症学会理事.
研究テーマと抱負タンパク質分解研究を軸足として主にアルツハイマー病の研究を行っている.抗体医療を超える創薬を目指したい.
ウェブサイトhttp://www.riken.jp/research/labs/cbs/proteol_neurosci/
論文リストhttps://www.ncbi.nlm.nih.gov/myncbi/takaomi.saido.1/bibliography/public/
趣味読書.