真核細胞内のさまざまな細胞小器官(オルガネラ)には固有のタンパク質が存在し,それらが機能することで,細胞に必要な物質とエネルギーが産生されている.種々のオルガネラは,独自の機能を発揮するとともに,オルガネラ間で情報を交換し,連携して細胞の維持,増殖,分化をなしとげている.このオルガネラ間の情報交換には,小胞輸送に加えて,異なるオルガネラが直接接触する場である“オルガネラ接触場”が重要な役割を担っている.特にミトコンドリアと小胞体の接触領域(mitochondria-ER contact sites:MERCs)は,効率的なカルシウムの受け渡しや脂質代謝の制御に重要な役割をすることが知られていたが,近年,MERCsがミトコンドリア分裂やアポトーシスを制御していること,オートファゴソームの形成場であること,細胞増殖や代謝調節のシグナル伝達の場であることなどの驚くべき知見が次々と発表され,MERCsの新たな機能が注目されている1).
さらに,MERCsの過形成により生じるミトコンドリアストレスが,アルツハイマー病や筋萎縮性側索硬化症(ALS)の病態形成に寄与していることや2, 3),ウイルスタンパク質によるMERCs形成阻害が抗ウイルス免疫応答を抑制することが報告され4),MERCs形成異常による病態形成という新たなパラダイムが確立されつつある.このようにMERCsは医学分野における病態の理解に重要であり,新たな創薬開発の標的としても注目されている.
筆者らはミトコンドリア外膜に局在するE3ユビキチンリガーゼであるMITOLを発見し,MITOLがミトコンドリアの形態調節などミトコンドリアの機能調節において重要な役割を果たしていることを報告してきた5, 6).さらにMITOLはミトコンドリアに会合する小胞体膜画分,すなわちMAM(mitochondria-associated membrane)画分に検出されることから,MERCsで機能することが想定された.MITOLの基質の一つとして同定されたミトコンドリア外膜の融合因子Mitofusin2(Mfn2)は,ミトコンドリアと小胞体の両オルガネラに局在し,互いのMfn2が重合することでMAMの係留因子として働くことが報告されていた.そこでMfn2によるMAM形成への影響を調べたところ,MITOLによるMfn2のユビキチン化がMfn2の重合化を誘導し,MAM形成を促進することを世界で初めて明らかにした7).また,マウスの脳を用いてMERCsを3次元的に観察することにも成功し,MITOLの欠損が海馬神経細胞のMERCsを減少させることを見いだし,生体内においてMITOLがMERCs形成を制御していることを示した8).さらに,MITOLのMAM領域における基質として,小胞体に局在する小胞体ストレス応答(UPR)センサー分子の一つであるIRE1αを同定した.解析の結果,MITOLはMAM領域を介してIRE1αをユビキチン化することにより,小胞体ストレスにより惹起されるIRE1αの過剰活性化を抑制して細胞死の実行を阻害していることを明らかにした9).このことはMAM領域が小胞体ストレス応答の調節の場になっていることを示している.本稿では,最近筆者らが見いだしたMERCsの新たな役割と,可逆的なMERCsの新規定量手法を概説する.
先述のとおり,細胞内に存在するさまざまなオルガネラは,互いにコミュニケーションをとるために近接し機能的な領域(オルガネラ接触場)を形成している.
オルガネラ接触場の定義にはさまざまな表現があるが,いくつかの総説では「相互作用する膜領域の近傍に空間的に限定された,生化学的に異なる分子組成を持つ領域」とされている11).また,オルガネラ接触場は,偶然近接したオルガネラ膜の相互作用ではなく,それぞれのオルガネラ膜に発現する係留因子どうしが,タンパク質-タンパク質相互作用することで形成される.
ミトコンドリアと小胞体の近接領域は,1959年に電子顕微鏡画像によって観察されており12),1990年に生化学的な手法によってMAMの単離・精製法が報告された13).2000年代に入るまでは網羅的な解析手法に乏しく,オルガネラ接触場の解析アプローチは,電子顕微鏡画像による2次元での解析や,密度勾配遠心法を用いた精製技術の向上に委ねられてきた.その後プロテオミクス技術の発展に伴い精製されたMAM画分に含まれるタンパク質の網羅的解析が行われて,さらに最近では蛍光タンパク質や顕微鏡技術の発展によって,より応用的かつ包括的なMERCsの機能解析が可能となりつつある11).
MERCsで機能するタンパク質,いわゆるMERCs関連因子はこれまでに複数報告されている.MFN2, IP3R, PDZD8などはその発現を抑制することによってMERCsの形成量が減少することから,MERCs係留因子として同定されている.形成されたすべてのMERCsが連動して機能するわけではなく,それぞれのMERCs係留因子によるMERCs形成機構には未解明な点も多い.
MERCsは飢餓刺激条件下でのラットの組織で観察されており,培養細胞においても飢餓誘導時に形成が促進される.特定のMERCs係留因子はグルコース飢餓条件で相互作用が増加し,MERCs係留因子の発現抑制によってミトコンドリア呼吸が低下することも報告されている.また,生理的な環境下でのMERCsは,恒常的なミトコンドリアでのATP産生のために重要な役割を担うことが示唆されている14, 15).したがって,細胞内要求度に応じた適切なMERCsの形成制御機構の存在が示唆されるが,その詳細はいまだ不明瞭であり,MERCsの形成・崩壊に関する詳細な分子機構の解明が必要である.
MERCsの主な機能の一つとして,ミトコンドリアへの物質輸送が報告されている.MERCsで輸送される物質として,カルシウムや脂質が明らかとなっており,カルシウム輸送を介したミトコンドリア動態や細胞死の制御への関与が複数報告されている16–18).
ミトコンドリアへ輸送されたカルシウムは,生理的環境下において酵素の補因子として機能するが,時には過剰な流入によって細胞死を誘導する引き金としての機能を発揮する.TCA回路では,ピルビン酸デヒドロゲナーゼ・イソクエン酸デヒドロゲナーゼ・α-ケトグルタル酸デヒドロゲナーゼがカルシウム依存的に活性化されるため,ミトコンドリア内でのATP産生において適切なカルシウム濃度の維持は不可欠である19).
一方で,MERCsを介した過剰なカルシウムの流入はミトコンドリア膜透過性遷移孔(mitochondrial permeability transition pore:mPTP)の開口を誘導し,Caspaseファミリーの活性化によるアポトーシスを誘起する.ミトコンドリアへの過剰なカルシウム流入は,MERCs係留因子の発現を抑制することで阻害でき,アポトーシスが抑制される20–22).
このように,MERCsを介したミトコンドリアへのカルシウム輸送は,定常時のミトコンドリア機能維持に不可欠である一方で,細胞死決定にも重要な役割を担っている.
空間的に限定された領域であるMERCsでは,他にもミトコンドリア機能維持に重要なさまざまな物質を安全かつ効率的にミトコンドリアへ輸送するための役割があることが予想される.筆者らは,後述するようにMERCsにおける鉄供給システムの存在を新たに見いだしており,今後もMERCsにおける未知の物質輸送・交換システムの発見に期待がかかる.
3. MITOL(mitochondrial ubiquitin ligase)
MITOL(別名:MARCHF5, MARCH5, RNF153)は,ミトコンドリア外膜に4回膜貫通して存在し,ユビキチン化を介して基質となるタンパク質を制御するE3ユビキチンリガーゼとして2006年に同定された23).
MITOLによってユビキチン化される基質はこれまでに20以上同定されており,その生理的意義はミトコンドリア動態,変性タンパク質の除去,炎症応答など多岐にわたることが報告されている.さらにMITOLは,アルツハイマー病やパーキンソン病,心不全などの疾患との関連も報告されており,ミトコンドリア外膜上でのMITOLによる基質へのユビキチン化修飾の重要性が注目されている24–26).
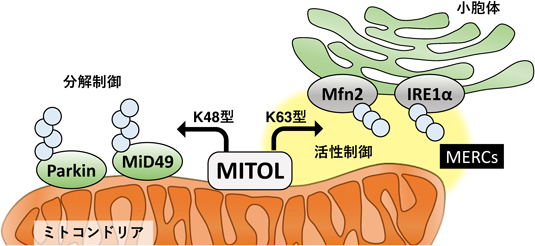
翻訳後修飾の一つであるユビキチン化にはさまざまな機能があり,その違いは基質に対して付加されるポリユビキチン鎖の形成様式によって異なる27).MITOLによってユビキチン化される基質への制御機構は主に2種類に大別される.主なものは,基質をプロテアソーム系による分解機構へと誘導するためのシグナルとして付加される“基質分解型”の制御である.“基質分解型”の制御を受ける基質に付加されるポリユビキチン鎖は,ユビキチンタンパク質の48番目のリシン残基(K)を介して形成される様式で「K48型」と称される.一方で,基質の発現量を変化させず,その機能を制御する“活性制御型”の修飾を受ける基質も複数同定されている.こちらの制御を受ける基質に付加されるポリユビキチン鎖は,2013年に同定されたMfn2をはじめ「K63型」が多く報告されている.これまでに報告されてきたMITOLの基質と,付加されるユビキチン鎖,およびその制御機構について,一覧にまとめて紹介する(表1).それぞれの制御機構に関する詳細や生理的意義については長島らの総説を参照されたい28, 29).
表1 MITOLの基質に付加されるユビキチン鎖と制御機構一覧| 基質 | ユビキチン鎖 | 制御機構 | 報告年 | 文献 |
|---|
| Fis1 | — | プロテアソーム分解 | 2006 | 23 |
| Drp1 | — | プロテアソーム分解 | 2006 | 47 |
| 2022 | 26 |
| mSOD1 | — | プロテアソーム分解 | 2009 | 48 |
| polyQ | — | プロテアソーム分解 | 2011 | 49 |
| TANK | K63型 | 活性制御 | 2011 | 50 |
| MAP1B-LC1 | — | プロテアソーム分解 | 2012 | 51 |
| Mfn2 | K63型 | オリゴマー形成 | 2013 | 7 |
| Mfn1 | K48型 | プロテアソーム分解 | 2014 | 52 |
| Prkar1a | K63型 | 活性制御 | 2015 | 53 |
| MiD49 | K48型 | プロテアソーム分解 | 2015 | 54 |
| MAVS (VISA) | K48型 | プロテアソーム分解 | 2015 | 55 |
| 2017 | 56 |
| 2020 | 57 |
| MCL1 | — | プロテアソーム分解 | 2016 | 58 |
| FUNDC1 | — | プロテアソーム分解 | 2017 | 59 |
| IRE1α | K63型 | オリゴマー化抑制 | 2019 | 9 |
| HBx | — | プロテアソーム分解 | 2019 | 60 |
| Parkin | K48型 | プロテアソーム分解 | 2021 | 25 |
| PolγA | K6型 | プロテアソーム分解 | 2021 | 61 |
| PEX3 | — | プロテアソーム分解 | 2022 | 62 |
| オートファジー分解 |
| PMP70 | — | ペキソファジー抑制 | 2022 | 62 |
| RMDN3 | K63型 | 活性制御 | 2022 | 31 |
| STING | K63型 | 凝集体形成抑制 | 2023 | 63 |
| MIC60 | K48型 | プロテアソーム分解 | 2023 | 64 |
| NLRP | K27型 | オリゴマー形成 | 2023 | 65 |
| BNIP | K48型 | プロテアソーム分解 | 2024 | 66 |
| DNA-PKcs | — | プロテアソーム分解 | 2024 | 67 |
| γc | K27型 | オートファジー分解 | 2024 | 68 |
| P53 | K48型 | プロテアソーム分解 | 2024 | 69 |
K48型のポリユビキチン鎖が付加され,ユビキチン・プロテアソーム系による分解制御を受ける基質の代表例として,MiD49, MAVS, Parkinなどが同定されている.これらの基質に対するユビキチン化は,そのほとんどがミトコンドリア外膜上で行われており,ミトコンドリアの動態制御・品質管理・シグナル伝達など,ミトコンドリアを起点とした反応を制御する共通点がある.
一方でK63型のポリユビキチン鎖が付加される基質として,Mfn2, IRE1α, RMDN3などが同定されており,これらの基質にはMERCsを介して機能が制御されるという共通点がある.
このようにMITOLは,K48型のユビキチン化修飾による基質の量的制御だけではなく,K63型のユビキチン化修飾を介したMERCsの機能制御において重要な機能を担うタンパク質であることが示された(図1).
MITOLによって基質に付加されるユビキチン鎖の選択機構についてはいまだ明らかとなっていないが,これまでに報告された例を踏まえると,MITOLが基質と相互作用する細胞内領域によって制御されている可能性が考えられる.特にMERCsは,他の細胞内領域とは異なる環境にあることから,MERCs独自のpHや酸化還元環境変化によってMITOLの活性が制御されていることも示唆される30).
また,最近ではMITOLがK6型・K27型のポリユビキチン鎖を修飾することや,プロテアソームを介さない基質の分解を制御することなども報告されており,MITOLが関与する機構の多様性が拡大し続けている.MITOLによる基質の選択性や,制御機構に関する詳細なメカニズムについて,その時間的・空間的な理解を含めたさらなる解析が期待される.
4. MITOLの新規基質・MERCsの新規機能の探索
これまでに報告されてきたMERCsにおけるMITOLの役割は,MERCsに局在する既知のタンパク質に対するユビキチン化を介した機能調節であった.一方で,MITOLは単なるMERCs関連因子の一つではなく,現時点でMERCsで機能する唯一のユビキチンリガーゼであることから,MITOLの近傍に存在する因子を網羅的に解析することにより,MITOLの新規基質のみならず,MERCsの新たな生理的機能の発見が期待された.
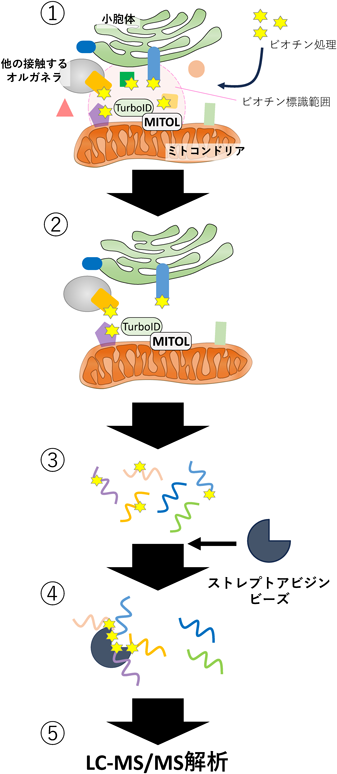
筆者らは,近位依存性ビオチン化酵素(APEX2またはTurboID)を融合したMITOLを発現する細胞を作製し,MITOL近傍因子を網羅的にビオチン化標識した.特にオルガネラ接触場に関する因子の同定を目的としていたため,ジギトニンを用いたアッセイによってサイトゾル成分を除き,膜タンパク質のみを含むサンプルを回収した.ストレプトアビジンビーズを用いてビオチン化タンパク質を精製し,質量分析法を用いて解析した.結果,既知のMITOLの基質を含む,さまざまなオルガネラの膜タンパク質が同定された(図2).特にその中でも,小胞体局在タンパク質として報告されている候補分子は,MERCsを介したMITOLによる制御を受ける基質である可能性が示唆された31).
5. MERCsを介したミトコンドリアへの鉄供給機構の発見
先述の手法により解析したMITOL近傍タンパク質として,ヘム分解酵素(heme oxygenase:HMOX)のアイソザイムの一つであるHMOX2が,既知のMERCs関連因子と同様に高い頻度で検出された.HMOXは1回膜貫通で小胞体に局在する,細胞内で唯一のヘム分解酵素であり,ヒトでは二つのアイソザイム(HMOX1, HMOX2)の発現が認められている32, 33).HMOXにヘムが結合すると,ヘムは鉄(二価鉄)・一酸化炭素・ビリベルジンに分解される.HMOX1は,過剰なヘムの蓄積や酸化ストレスに応答して発現量が増加される誘導型酵素であり,抗酸化作用をもたらすことが知られている34–36).一方で,今回MITOLの近傍因子として同定されたHMOX2はHMOX1のようなストレス応答性を持たない構成(常在)型酵素として知られているため,ストレス非依存的なHMOX2独自の活性がMERCsで機能することが示唆された.
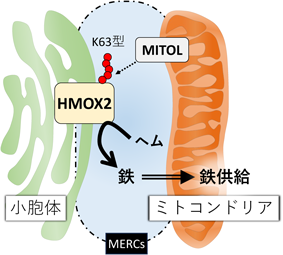
そこで,密度勾配遠心法を用いた細胞分画を行い,HMOX2の細胞内発現領域の特定を試みたところ,HMOX2は確かに小胞体画分およびMAM画分で検出された.また,免疫沈降法を用いてHMOX1とHMOX2に対するユビキチン化を確認したところ,MITOLはHMOX1をユビキチン化せず,HMOX2を特異的にユビキチン化することが明らかとなった.MITOLによるHMOX2のユビキチン化は,MERCs係留因子の発現抑制時に減弱することから,MERCsを介した制御であることも確認された.さらに,MITOLによってHMOX2に付加されるポリユビキチン鎖の形成様式はK63型であり,MERCsを介してMITOLによる制御を受ける既知の基質と一致した.これらの知見から,MITOLによるHMOX2のユビキチン化が,MERCsを介したHMOX2の機能制御であることが示唆された.
HMOXが細胞内で唯一のヘム分解酵素であることを踏まえ,HMOX2のヘム分解活性を細胞内で可視化する実験系を構築し,MITOLによるユビキチン化が与える影響を評価した.その結果,MITOLによってユビキチン化されるリシン残基を変異させたHMOX2では,野生型と比較してヘム分解活性が低下することを観察したため,MITOLによるユビキチン化はHMOX2のヘム分解活性を制御することが見いだされた.
次に,MERCsを介したMITOLによるHMOX2活性制御機構の生理的意義を同定するため,ヘムの分解によって産生される鉄とミトコンドリアの関係性に着目した.
細胞内の鉄はミトコンドリアに豊富に存在し,鉄硫黄クラスターやヘムの合成に利用されることから,ミトコンドリアは細胞内の鉄代謝中枢として機能する37).ミトコンドリアで合成された鉄硫黄クラスターやヘムは,さまざまなタンパク質の活性部位に配位されることで,電子伝達や酸素運搬などの機能を獲得する.特にミトコンドリアの呼吸鎖I~IVには,活性部位にヘムを持つシトクロムや鉄硫黄クラスターが複数存在し,電子伝達の中心的な役割を担っている.ミトコンドリア鉄欠乏時には,呼吸鎖の崩壊やミトコンドリア呼吸およびATP合成が低下することから,ミトコンドリアの機能維持において鉄は必要不可欠である38–41).そのため,細胞内の鉄は効率的にミトコンドリアへ輸送される必要があるが,ミトコンドリアへの鉄輸送機構についてはいまだ詳細が不明瞭であり,特にヘム分解によって産生された鉄がミトコンドリアに与える影響は明らかとなっていない.
HMOX1とHMOX2は,どちらもヘム分解産物として鉄を産生するが,ヘム過剰時には,鉄貯蔵タンパク質であるフェリチンの発現も誘導されることから,HMOX1によるヘム分解産物の鉄はフェリチンによって貯蔵されることが予測できる42).一方で,恒常的に発現するHMOX2のヘム分解によって産生された鉄の行方はいまだ明らかとなっていないが,最近の報告では,HMOX2が細胞増殖に十分な鉄供給源として機能することが示された43).
先述のとおり,MERCsではミトコンドリア恒常性維持に必要なカルシウムや脂質の輸送が行われていることから,MERCsにおけるHMOX2のヘム分解が,ミトコンドリアへの鉄供給機構として機能することが示唆された.
HMOXによるヘム分解がミトコンドリアに与える影響を評価するため,ヒト子宮頚がん由来細胞株(HeLa)を用いて,HMOX1ノックアウト細胞,HMOX2ノックアウト細胞,HMOX1・HMOX2のダブルノックアウト細胞を作製した.その後,作製した各細胞からミトコンドリアを単離し,ICP-MS解析によって含まれるイオン濃度を定量した.その結果,HMOX1ノックアウト細胞では,野生型の細胞と比較して顕著な変化は見受けられなかったが,HMOX2ノックアウト細胞とダブルノックアウト細胞では,ミトコンドリア内の鉄量が顕著に低下していることを見いだした.さらに,HMOX2ノックアウト細胞でみられたミトコンドリア鉄量の低下は,野生型のHMOX2を再発現することによって回復したが,酵素活性部位のアミノ酸を変異させたHMOX2(ヘム分解不活化変異体)の再発現では回復しなかった.これらの結果から,ミトコンドリア内の鉄量はHMOX2のヘム分解によって維持されていることが明らかとなった.
次に,MERCsを介したMITOLによるHMOX2へのユビキチン化・ヘム分解制御がミトコンドリアへの鉄供給を調節する機構であることが予測されたため,MITOLによってユビキチン化されないHMOX2変異体を再発現させた細胞を作製し,ミトコンドリア内の鉄量を測定した.その結果,野生型のHMOX2を再発現することによって回復するミトコンドリア内の鉄量は,MITOLによってユビキチン化されないHMOX2変異体を再発現しても回復しないことが確認された.よって,MITOLによるHMOX2のユビキチン化のユビキチン化は,ミトコンドリアへの鉄供給を調節する機構であることが明らかとなった.
また,ミトコンドリア内の鉄は,鉄硫黄クラスターやヘムに合成された後,呼吸鎖の構成因子に配位され,電子伝達を担うことから,ミトコンドリア呼吸を制御することが予測された.フラックスアナライザーを用いた酸素消費速度(OCR)の測定を行った結果,HMOX2ノックアウト細胞ではOCRが顕著に低下し,野生型のHMOX2を再発現することによって回復する一方で,MITOLによってユビキチン化されないHMOX2変異体を再発現しても回復しないことが確認された.したがって,MITOLによるHMOX2のユビキチン化は,ミトコンドリア内鉄量と同様に,ミトコンドリア呼吸の維持にも重要であることが明らかとなった.
鉄は,ミトコンドリア呼吸などに必要不可欠である一方,ミトコンドリア内の過剰な鉄蓄積は活性酸素種(reactive oxygen species:ROS)の産生を誘導することから,ミトコンドリア内では厳密な鉄恒常性の制御が必要である.MITOLによるHMOX2のユビキチン化は,ミトコンドリアへの鉄供給量を制御する機構であることから,ミトコンドリア内の鉄要求度に応じて,MITOLがHMOX2のヘム分解活性を適切に調節していることが予測された.
MITOLによるHMOX2へのユビキチン化が増減する条件を探索した結果,細胞内のヘム量変化には応答せず,細胞内・ミトコンドリア内鉄欠乏を模倣する刺激を加えることによって促進することを見いだした.以上の知見から,MITOLによるHMOX2へのユビキチン化は,ミトコンドリア内鉄欠乏に応答したヘム分解を調節し,ミトコンドリア鉄供給制御を介したミトコンドリア恒常性の維持機構として機能することが示唆された(図3).
MITOLによるMERCsを介したHMOX2の制御機構は,これまでブラックボックスとされてきた「ミトコンドリアへの効率的な鉄供給機構の解明」に限らず,「MERCsにおける新規機能の同定」および「HMOX2によるヘム分解を介した鉄供給の重要性」など複数の分野における基盤研究としてそれらのさらなる理解に貢献する.
しかしながら,ミトコンドリア内鉄恒常性がMERCsにおいて調節される意義や,ミトコンドリア内鉄欠乏シグナルの伝播機構は未解明であり,MERCs独自の特徴や細胞内における鉄の毒性など,さまざまな要因との関与を踏まえた解析が必要となる.また,HMOX2のヘム分解反応は,鉄の他に一酸化炭素やビリベルジンを産生し,どちらも抗酸化作用があることが報告されている34–36).HMOX2のヘム分解制御を介した一酸化炭素やビリベルジンの量的制御と,MITOLによるHMOX2のユビキチン化の関係性についてはさらなる研究が必要である.
ここで,筆者らが最近開発したハイスループットかつ生細胞で可逆的なオルガネラ間接触を定量可能なツールの紹介と,その応用について言及しておきたい.
前述のとおり,オルガネラ間の接触は単なる物理的接触ではなく,必要に応じて係留因子の制御を介した接触場形成量が調節されている.また,オルガネラ接触領域が作り出す特殊な環境は,接触と解離を繰り返すことで高いモビリティを持つことも特徴的である.そのため,MERCsの多彩な機能・役割を理解するには,いかに時空間情報を維持したまま解析し,適切なオルガネラ接触の情報を得られるかが鍵となる.
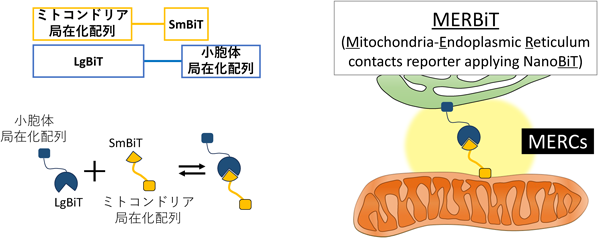
現在,電子顕微鏡(EM),Split-GFP, SPLICS, PLA法,hypotonic bufferを使用したオルガネラ間接触の測定系が一般的に用いられている44, 45).我々もこれらの系を用いて解析を行ってきたが,いずれの手法も不可逆的にオルガネラ間接触を誘導してしまうことや細胞を固定しなければならないなど実験上の制約があり,時空間的情報を失った状態で測定せざるをえなかった.そこで我々はNanoBiTに着目し,生細胞で可逆的なオルガネラ間接触を定量可能なツールの開発に着手した.
NanoBiTはNanoLucルシフェラーゼをベースにしたシステムで,生細胞内でのタンパク質間相互作用が高感度に発光検出できる.NanoBiTはLargeBiT(LgBiT)およびSmallBiT(SmBiT)の小さな二つの分子で構成されており,LgBiTとSmBiTが結合し発光する.他の多くのシステムと異なりNanoBiTは可逆的で速やかに解離すること,また細胞透過性のある発光基質を使用することから,リアルタイムでの可逆的な細胞内分子相互作用の検出に適している.この技術を応用し,LgBiT, SmBiTのそれぞれにオルガネラ局在化配列を付加することで,生細胞における可逆的なオルガネラ間接触を定量することができると推測した.実際にミトコンドリア局在化配列-LgBiT, SmBiT,小胞体局在化配列-LgBiT, SmBiTを作製し,細胞へ導入後,既知のミトコンドリア-小胞体係留因子の発現を抑制するとその発光量が減衰した.また,細胞をHBSS(細胞飢餓誘導)培地で飼育するとMERCsが増加することが知られていることから46),細胞をHBSS培地で飼育し発光値を計測したところ発光値は増加し,HBSS培地で飼育したのちに通常培地に戻すと発光値は減衰した.このことから,生細胞を用いて可逆的なMERCsが定量可能であると判断した.このシステムをMERBiT(Mitochondria-Endoplasmic Reticulum contacts reporter applying NanoLuc Binary Technology)と名づけた(図4).
さらに我々はMERBiTシステムを用いた細胞にストレスを与え,MERCsが増加・減少する未知の条件をスクリーニングした.その結果,ミトコンドリア呼吸鎖I, IIIの阻害剤であるRotenoneやAntimycin Aを細胞に添加した際,MERCsが有意に増加することを発見した.また,ミトコンドリアATP産生を阻害するミトコンドリア呼吸鎖V阻害剤Oligomycinの添加ではMERCsが増加しなかったことから,ATPの減少はMERCsの増加を誘導しないと結論づけた.RotenoneやAntimycin AはミトコンドリアATP産生低下を引き起こす一方でROSの産生を促進させることが報告されている.よって,RotenoneやAntimycin Aによって誘導されたMERCsはROSによる影響ではないかと考え,ミトコンドリア特異的ROS除去剤であるMito-TEMPOを添加したところMERCsの増加が抑えられた.このことから,ミトコンドリアROSがMERCsを増加させることが明らかとなった.
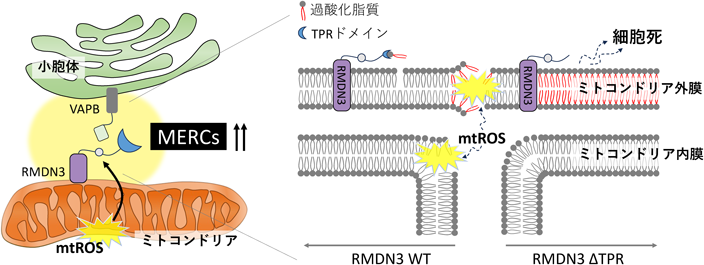
次にROSにより誘導されるMERCsがどの因子によって係留されているかを調べるため,MERBiTシステムを用いて既知の係留因子を発現抑制し解析を行った.結果,RMDN3(ミトコンドリア局在)やVAPB(小胞体局在)を発現抑制した際にROS誘導性のMERCs形成が減少した.このことからROS誘導性のMERCs形成にはRMDN3-VAPBの係留が必要であることが明らかとなった.
さらに筆者らは,RotenoneやAntimycin A刺激の際,RMDN3やVAPBを発現抑制すると,ミトコンドリアに過酸化脂質が過剰蓄積することや細胞死が亢進することに気がついた.そこで,ROS誘導性MERCs形成の細胞保護機能について解析することにした.
興味深いことに,RMDN3は脂質転移ドメインであるTPR(tetratricopeptide repeat)を持ち,脂質転移活性を有する.そこで,RMDN3のTPRドメインが毒性の高い過酸化脂質を転移させ,細胞死を抑制している可能性を疑った.結果,RMDN3のTPRは過酸化脂質の転移能を持つこと,またRMDN3が過酸化脂質を小胞体に転移させミトコンドリア過酸化脂質の毒性を希釈することで細胞死を抑制していることを明らかとした(図5).さらに,これらの分子機構が熱産生組織である褐色脂肪細胞にも保存されていることも発見した.
これまでオルガネラ接触解析は,測定法が煩雑であるがゆえ思うように行えなかった.前述の研究成果は,MERBiTシステムの開発に成功し,可逆的なオルガネラ接触を簡便かつ迅速に解析できるようになった結果であると考える.現在はMERBiTシステムの成功例に倣い,ミトコンドリアをハブにしたいくつかのオルガネラ接触計測系の開発を行っている.今後,さらなるオルガネラ接触の生理的役割の発見に期待がかかる.
マウスの各臓器におけるMITOLの発現量は加齢とともに顕著に減少することから,加齢によるミトコンドリアの機能異常は,MITOLの発現低下が原因の一つであると考えられる.実際に心臓特異的MITOL欠損マウスはβ-ガラクトシダーゼ染色領域の拡大,広範なリポフスチンの沈着と線維化の亢進など心筋老化を示し,最終的に心不全に至る26).脳特異的MITOL欠損マウスではアルツハイマー病やパーキンソン病の病態が増悪することを報告している24, 25).さらに,ケラチノサイト特異的MITOL欠損マウスは,生後6か月ごろから顕著な白髪・脱毛・皮膚炎症が認められ,病理組織解析においても皮脂腺の増加・表皮の肥厚など皮膚の老化所見が観察されている(論文投稿中).このようにMITOLの各臓器欠損マウスはそれぞれの臓器の老化を反映しており,これらの老化モデルマウスを解析することにより,老化の分子機構の解明と予防法開発につながると期待している.
さらに筆者らはMITOLの発現を誘導する薬剤は老化を抑制できるのではないかと考え,そのような活性を持つ薬剤を漢方薬ライブラリーから探索した結果,オウレンやオウバクの抽出液がMITOLの発現を上昇させることを見いだした.それらの共通成分であるベルベリンそのものにはMITOLの発現を上昇させる活性はなかったが,ベルベリンの代謝物XがMITOLの発現を誘導することがわかった.筆者らは代謝物Xに種々の化学修飾を加え,活性の強い新規物質の合成にも成功している(特許出願中).この物質を含め,ミトコンドリアを活性化する代謝物Xの誘導体群を総称してマイトルビンと命名した.現在,マイトルビンをさまざまな分野の老化研究の専門家に送って抗加齢活性を検証してもらっている.これまでに予想を超えるよい報告を受けているので,近い将来,マイトルビンがミトコンドリア病をはじめ多くの加齢性疾患に苦しむ患者を救う薬になるのではないかと期待に胸を膨らませている.
最後にMERCsを標的にした創薬開発にもふれておきたい.上述したように,筆者らはNanoBiTシステムを応用し,ハイスループットかつ生細胞で可逆的なMERCsを定量可能とした新規ツールであるMERBiTシステムの開発に成功した.実際にMERBiTシステムを用いてミトコンドリア障害におけるMERCsの生理的重要性を明らかにした(Nat. Commun. in revision).このMERBiTシステムを用いて世界初のMERCsを標的にした創薬スクリーニングが可能である.マイトルビンを超える新たなミトコンドリア活性化薬が見つかるかもしれない.共同研究にてMERBiTシステムを提供しますので,ご興味のある方はぜひ連絡いただきたい.