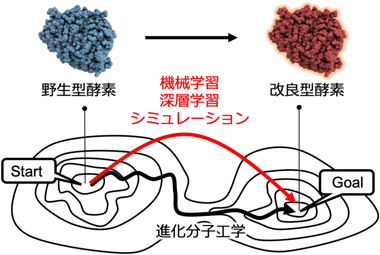
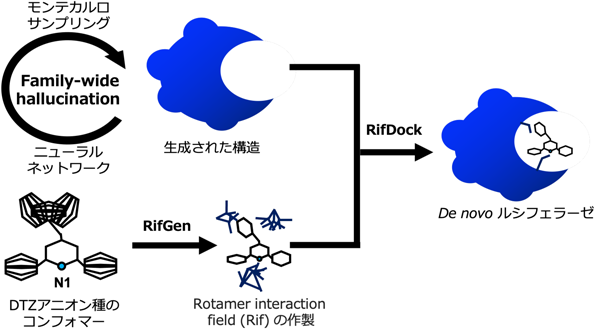
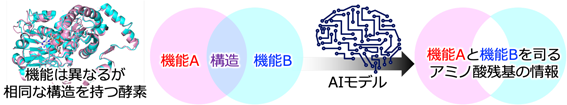
代謝酵素の基質認識に寄与するアミノ酸残基の推定と機能創出Estimation and function creation of amino acid residues contributing to substrate recognition in metabolic enzymes
大阪大学大学院情報科学研究科バイオ情報工学専攻Department of Bioinformatic Engineering, Graduate School of Information Science and Technology, Osaka University ◇ 〒565–0871 大阪府吹田市山田丘1–5 情報科学研究科B棟6F ◇ 1–5 Yamadaoka, Suita, Osaka 565–0871, Japan
発行日:2024年10月25日Published: October 25, 2024