ヒトの脳の重量は体重の2%程度であるが,エネルギー消費量は体全体の20%を占める.このような脳の大量のエネルギー消費の相当部分はタンパク質合成に使われる.タンパク質合成は,脳ではたとえば長期記憶の形成に必須であり,個体レベルでは,翻訳阻害剤であるピューロマイシンをマウス脳に作用させた際に記憶が形成されないことが1960年代から知られていた.細胞レベルの記録機能においても,神経細胞どうしをつなぐシナプスに高頻度に刺激が来た後にシナプスの伝達効率が変化するシナプスの可塑性には,タンパク質合成が必須である.
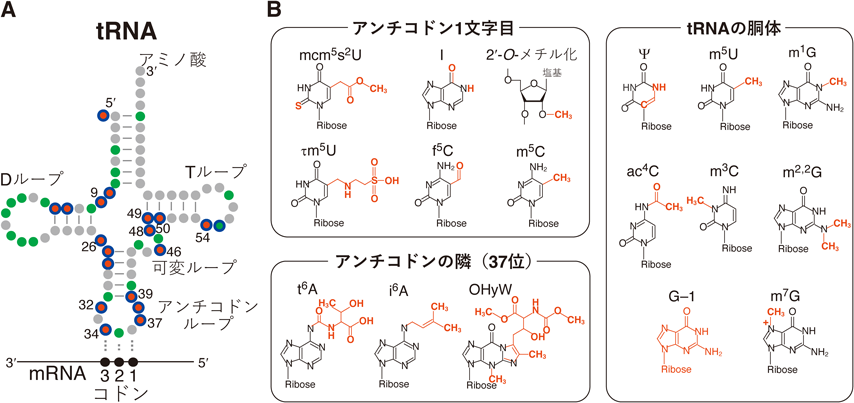
タンパク質合成において,tRNAはコドンとアミノ酸をつなぐアダプター分子として働く(図1A).ヒトゲノムにはtRNA遺伝子が500種類以上存在し,通常はそのうち200種類ほどが転写される.転写されたtRNAはクローバーリーフ様の二次構造をとり,DループとTループを重ねあわせてL字型の立体構造をとる.tRNAはリボソームに入りmRNAのコドンを解読してアミノ酸を新生ペプチド末端に受け渡すため,tRNAのアンチコドンループから3′末端のアミノ酸までの距離は,リボソームの暗号解読中心とペプチド転移活性中心の距離に相当する約70 Åとなっている.つまり,数百種類のtRNAはその配列の多様性にかかわらずほぼ同一の立体構造をとるという制約を課されている.このようなtRNAの立体構造の形成と精確なコドン解読は,tRNAの転写後修飾(tRNA修飾)により支えられている1).tRNA修飾は,メチル化や水素付加のような単純な化学構造から,アミノ酸付加やワイブトシン修飾のような複雑な化学構造まであり(図1B),ヒトでは合計40種類以上のtRNA修飾が知られる.tRNAは非常に高密度に修飾を受けており,一般的な76塩基ほどの長さのtRNA分子には,13個前後の修飾が存在する.tRNA修飾は全身の組織で高効率に入れられており,脳をはじめとする神経組織はtRNA修飾に特に強く依存している2).
tRNA修飾は特定の酵素によりtRNA中の特定の位置に導入される.ヒトにおいて,tRNA修飾を担うタンパク質(修飾酵素とそのパートナータンパク質)は,未同定のものも含めて100種類ほどあると見積もられる.これらのtRNA修飾を担うタンパク質のうち半分ほど(50種類以上)で疾患変異が見つかっており,tRNA修飾の生理的重要性を物語っている.RNA修飾の欠損が引き起こすさまざまな疾患は「RNA修飾病」と総称され1, 3),中でもtRNA修飾の欠損や異常が引き起こす疾患を「tRNA修飾病」と呼ぶことを我々は提唱している2).tRNA修飾病を疾患の種類で大別すると,脳・神経疾患,腎障害,低身長,ミトコンドリア病,がんが大部分を占める.tRNA修飾病の全体像の概要について本節で記すが,神経以外のtRNA修飾病の詳細は他の総説2)を参照されたい.
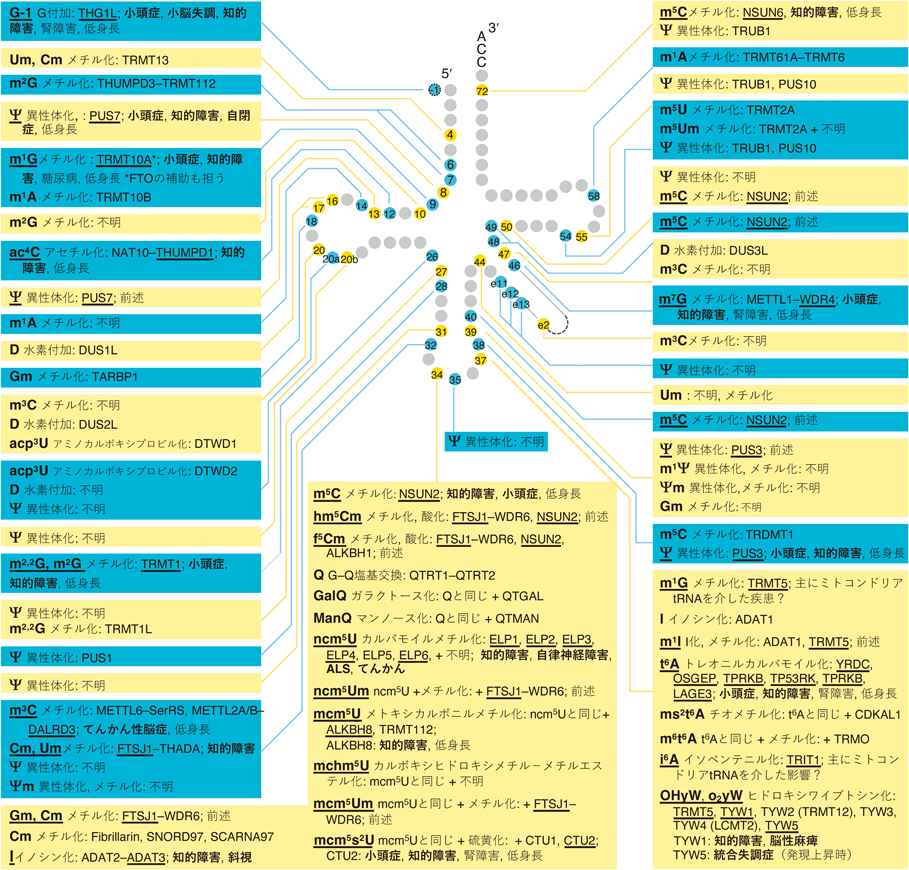
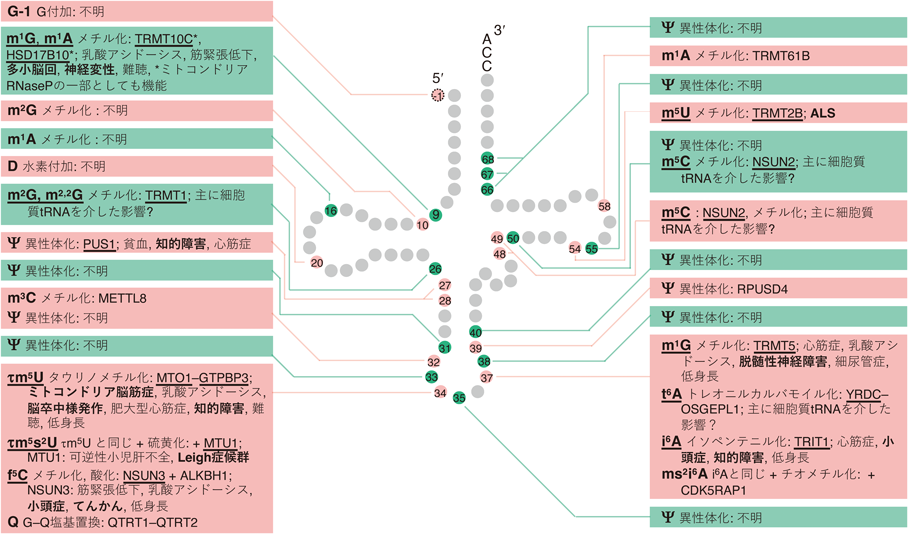
tRNA修飾病の中で,責任変異を有する修飾関連タンパク質の種類が最も多いのは,脳・神経疾患であり,現時点で36種類のタンパク質について責任変異が知られ,毎年のようにその数が増えている(図2,図3).これらの脳・神経疾患の多くは低身長(成長遅延)を伴い,一部は腎障害も併発する(図2,図3).逆に,低身長や腎疾患を発症するtRNA修飾病はほぼ必ず脳・神経疾患を伴う2).脳・神経のtRNA修飾病について,詳しくは次の節以降で述べる.
単一のtRNA修飾酵素遺伝子の変異で最も患者人口が多いのは2型糖尿病である.tRNA修飾酵素の一種をコードするCDKAL1遺伝子の特定の一塩基多型(SNP)は,2型糖尿病の罹患リスクを上げ,CDKAL1リスク型SNPによる2型糖尿病患者は日本だけで少なくとも数十万人いると見積もられる.CDKAL1タンパク質は,リシンtRNAの37位(アンチコドンの隣)に存在するms2t6Aという大きな修飾の中の2-メチルチオ化(ms2)修飾を担う酵素である.食事により血糖値が上がると膵臓β細胞においてインスリン前駆体が大量に合成され,その後切断されてインスリンが作られる.この際に,β細胞内での翻訳量の半分近くがインスリン前駆体の翻訳に費やされる.インスリン前駆体の切断部位のアミノ酸はリシンであり,CDKAL1の機能低下によりリシンtRNAのms2修飾が減少してリシンの翻訳を間違いやすくなると,β細胞で小胞体ストレスが上昇し,長期的にはβ細胞が疲弊して2型糖尿病が発症しやすくなる4).
ミトコンドリアの中には,太古に細胞内共生した好気性細菌に由来する16.6 kbpの環状DNAが核DNAとは独立に存在する.ミトコンドリアDNAには,呼吸鎖複合体を構成するタンパク質のうち13種類の遺伝子と,その翻訳に使われるリボソームRNAとtRNAの遺伝子がコードされている.ミトコンドリアtRNAは転写後に修飾を受けるが,修飾を担う20種類以上のタンパク質は核にコードされている.ミトコンドリア翻訳に影響が特に大きい修飾酵素の機能欠失変異は,呼吸鎖複合体の機能低下を引き起こし,好気的なATP合成の低下や,それに伴う解糖系の亢進による乳酸の蓄積,NADHの蓄積,NAD+の減少,活性酸素種の発生,ミトコンドリアへのタンパク質の輸送低下とそれに伴う細胞質でのタンパク質の凝集などが引き起こされる.結果,乳酸アシドーシスや特にエネルギー消費量の多い心臓や脳などの臓器に異常が生じる「ミトコンドリア病」が発症する.
がん細胞は細胞増殖が盛んなためタンパク質合成に強く依存し,さまざまなtRNA修飾酵素の発現増加ががん細胞の増殖を支えることが知られる.一方,種類は少ないがtRNA修飾の低下ががんの進行を促進する例も知られる.細胞質のフェニルアラニンtRNAのアンチコドンの隣(37位)に存在するヒドロキシワイブトシン修飾(OHyW,図1B)は,6種類のtRNA修飾酵素により段階的に形成される修飾である.その3段階目の反応を担うTYW2をコードする遺伝子のプロモーターメチル化による転写抑制の度合いが,さまざまながん,特に大腸がんの予後の悪さと相関する.分子レベルでは,TYW2の発現低下によるOHyW修飾の低下によりPheコドンでリボソームがコドンの読み枠を間違えやすくなり,それによりnonsense-mediated mRNA分解経路が発動しやすくなる.結果的に大腸がん細胞の増殖に有利なトランスクリプトームになり,がんの進行が促進される.
tRNA修飾酵素の変異により神経疾患が発症する修飾について,図2,図3ではtRNA修飾名とその酵素に下線を引き,神経関連の症状,および併発する他の症状を記載した.現時点で36種類の修飾関連タンパク質の変異が脳・神経の疾患を引き起こすことが知られる.そのうち26種類は主に細胞質tRNAの修飾を担うタンパク質であり,10種類は主にミトコンドリアtRNAの修飾を担うタンパク質である.
細胞質tRNAの修飾酵素の遺伝子変異が引き起こす神経のtRNA修飾病で最も多い症状は,知的障害と小頭症である.知的障害と小頭症は,いずれも脳の発生,発達の不全が引き起こす.小頭症は脳の発生と発達のプロセスの不具合により頭が異常に小さい状態であり基本的に知的障害を伴う.また,脳の発生と発達の不具合が比較的軽微な場合は,自閉症スペクトラム障害などの精神疾患が引き起こされる例がある.大まかには,脳の発生,発達の不全の度合いが強い順に,小頭症(と併発する知的障害)>知的障害>自閉症スペクトラム障害などの精神疾患が発症する.神経組織の発生,発達の不全は,興奮性神経細胞と抑制性神経細胞のバランスの異常などによりてんかんを伴うことも多い(図2).
ミトコンドリアtRNAの修飾のうちミトコンドリア翻訳に特に影響が大きい修飾を担うタンパク質の変異は,好気的なATP合成の低下とそれに伴う乳酸の蓄積,NADHの蓄積とNAD+の不足,活性酸素種の増加などにより,乳児期からミトコンドリア病を引き起こす.たとえばアンチコドン1塩基目の5-タウリノメチルウリジン修飾(τm5U,図1B)を担うMTO1とGTPBP3の変異や,5-ホルミルシチジン修飾(f5C,図1B)を担うNSUN3の変異は,患者により症状がやや異なるが乳酸アシドーシス,肥大型心筋症,成長遅延に加えて,脳幹の壊死や大脳萎縮,てんかんといった脳の障害を引き起こす.多くのミトコンドリア脳筋症患者でみられるミトコンドリアDNAのA3243G変異は,ロイシンtRNA(UUR)遺伝子内にあり,変異ロイシンtRNAはτm5U修飾を受けることができず,ミトコンドリア翻訳量が減少し,ミトコンドリア脳筋症を引き起こす.ミトコンドリアtRNA 9位のメチル化酵素TRMT10Cやそのパートナータンパク質HSD17B10の変異は,多小脳回や神経変性,乳酸アシドーシス等を引き起こす.ただし,TRMT10CとHSD17B10はミトコンドリアtRNAの5′末端切断酵素複合体の一部としても機能しており,メチル化活性と5′末端切断酵素活性のどちらの低下がどれだけ関与しているかは切り分けられていない.以上のように,影響の大きなミトコンドリアtRNA修飾酵素の機能欠失変異は,神経発生の障害と神経変性の両方を引き起こす.中枢神経だけでなく末梢神経についても,ミトコンドリアtRNAの54位に5-メチルウリジン修飾(m5U,図1B)を導入するTRMT2Bの変異による若年性の筋萎縮性側索硬化症(ALS)が知られる.
細胞質tRNAの修飾を担うタンパク質の変異による神経のtRNA修飾病の多くは,乳幼児から発症する小頭症や知的障害といった中枢神経の発生と発達の不全による疾患である.一部,末梢神経の疾患として細胞質tRNA 34位の5-メトキシカルボニルメチル2-チオウリジン修飾(mcm5s2U,図1B)修飾に必要なELP1の変異による自律神経障害やELP3の変異によるALSといった例も知られるが,数は比較的少ない.責任変異が報告されている神経のtRNA修飾病は基本的に遺伝性疾患であり,全身の組織が同じ変異を持つにもかかわらず,細胞質tRNA修飾の欠損の場合は,神経組織以外の大部分の組織,臓器に異常がみられないことも多い.現状では,疾患の表現型が神経組織に偏る原因は大部分が未解明である.一方,修飾酵素のノックアウトマウスを用いた最近の研究により,疾患の脳特異性の原因の一端が解明された例もあり後述する.遺伝性の神経のtRNA修飾病では,組織としての明瞭な機能異常は神経組織だけでみられることが多い一方で,多くの患者では,低身長(成長遅延)も併発する(図2,図3).現時点では,多くの神経のtRNA修飾病患者で低身長が併発することの具体的な原因は不明である.全身の細胞における翻訳低下や,脳で分泌され全身の成長に関わるホルモン(成長ホルモン,甲状腺刺激ホルモンなど)の量の減少といった可能性が考えられ,今後の解明が期待される.
tRNA修飾病の発症機構を理解するためには,tRNA修飾の分子機能の理解が不可欠である.tRNA修飾の主な機能は,(1)mRNAのコドンの精確な解読の実現と(2)tRNAの構造の維持と分解からの保護に大別される.
コドン解読に関わる修飾は,アンチコドン1塩基目(tRNAの34位)の修飾と,アンチコドンの隣(37位)の修飾である(図1).tRNAのアンチコドン1塩基目(34位)は,mRNAのコドン3塩基目と対合する.アンチコドン1塩基目がAの場合,コドン3塩基目としてUのみが対合できる.一方,アンチコドン1塩基目のAがイノシン化修飾(I,図1B)を受けると,コドン3塩基目としてA, U, Cの3種類と対合可能になる(塩基対合の拡張).同様に,アンチコドン1塩基目がCの場合,コドン3塩基目としてGのみと対合できるが,5-ホルミルシチジン修飾(f5C,図1B)を受けると,G, Aの2種類と対合可能になる(塩基対合の拡張).アンチコドン1塩基目がUの場合,リボソーム内ではコドン3塩基目として特別にA, U, G, Cが許容されるが,5-メトキシカルボニルメチルウリジン修飾(mcm5U)や5-タウリノメチルウリジン修飾(τm5U)を受けると,コドン3塩基目としてAとGのみが許容されやすくなる(塩基対合の制限).さらに,これらの修飾Uの2位に結合している酸素が硫黄に置換されたmcm5s2Uやτm5s2Uは,大きな硫黄原子の立体障害によりリボースの形がC3′エンド型をとりやすくなり,よりA, Gのみが許容されやすくなる.
tRNAのアンチコドンの隣(37位)の塩基修飾は,精確なコドン認識とコドンの読み枠の維持を支える役割を有する.たとえば,コドン1塩基目がAのコドン(ANNコドン,Nは任意の塩基)を解読するさまざまなtRNAの37位には,N6-トレオニルカルバモイル修飾(t6A,図1B)があり,コドン1塩基目のAの塩基とスタッキングすることで,コドン–アンチコドン対合を強化する.AAA/AAGコドンを解読するリシンtRNAと,UUU/UUCコドンを解読するフェニルアラニンtRNAの37位には,それぞれのtRNAにしか存在しないms2t6A修飾やOHyW修飾が存在する.これらの修飾は,mRNA配列がAAAAやUUUUのようなリボソーム内で読み枠を間違えやすい箇所において,読み枠の維持を支えると考えられている.本段落でふれたコドン解読に関わる修飾の多く(I, mcm5s2U, τm5U, t6A, OHyW)は,その責任酵素の機能欠失変異により小頭症,知的障害などの脳・神経疾患が発症する(図2,図3).
tRNAの立体構造の維持を担う修飾は,tRNAの全体に存在する.たとえば,tRNAの26位には,N2,N2-ジメチルグアノシン修飾(m2,2G,図1B)がある.m2,2G修飾のメチル基はG–Cの塩基対合を形成不能にする働きを有する.そのため,26位のGが未修飾だとtRNA中で本来対合してはいけないCと対合してtRNAが異常な全体構造をとる例が計算で予測されている.tRNAの9位に存在するN1-メチルグアノシン修飾(m1G,図1B)もG–Cの塩基対合を形成不能にする働きを有しm2,2Gと似た働きをする可能性が考えられるが,検証が待たれる.シュードウリジン修飾(Ψ,図1B)は,ウリジンのウラシル塩基を切り離し回転させてリボースに再結合させた異性体であり,tRNAのさまざまな位置に存在する.UがΨになることで塩基中の窒素が溶液側に露出して周囲と水素結合を形成可能になることで,結果的にリボースがC3′エンド型をとりやすくなり塩基がスタッキングしやすくなる.この作用により,27位,39位,50位のΨの存在がtRNAの立体構造の安定化(融解温度の上昇)を引き起こす例が知られる.本段落でふれた修飾(m2,2G, m1G, Ψ)についても,修飾酵素の機能欠失変異により小頭症,知的障害などの脳・神経疾患が発症する(図2).
tRNAを分解から守る修飾もtRNAの全体に存在する.なお,RNAのエキソヌクレアーゼの多くは二本鎖構造を有するRNAは分解しづらく,構造がゆるんだRNAを分解しやすいため,tRNA修飾によるtRNAの立体構造の維持と分解からの保護の間には密接な関係があると考えられる.tRNAの可変ループ内の46位にはN7-メチルグアノシン修飾(m7G,図1B)がある.哺乳動物でm7G修飾酵素METTL1が失われるとさまざまなtRNAが分解し,さらに酵母ではこの分解は5′-3′エキソヌクレアーゼRat1とXrn1によるrapid tRNA decay(RTD)経路が担うことが知られる.また,哺乳動物ではFTSJ1が多くのtRNAの32位と34位のリボース2′-O-メチル化修飾(図1B)を担い,フェニルアラニンtRNA(tRNAPhe)では32位と34位の2′-O-メチル化修飾がさらに37位のOHyW修飾反応を促進する.Ftsj1ノックアウトマウスでは脳でtRNAPheが分解し,この分解はエンドヌクレアーゼによるアンチコドンループの切断を介して開始すると考えられている.本段落でふれた修飾(m7G, 2′-O-メチル化,OHyW)についても,修飾を担うタンパク質の機能欠失変異により先天性の小頭症,知的障害,脳性麻痺といった,脳・神経疾患が発症する(図2).
以上のように,tRNA修飾は主にmRNAのコドンの解読やtRNAの立体構造の維持と分解からの保護を担う.また,これらの主要な機能に加えて,さまざまな修飾には自然免疫からtRNAが非自己RNAとして認識されないようにする役割があることと,一部のtRNA修飾はtRNAをアミノアシル化する酵素や他のtRNA修飾酵素の認識部位としての役割などを有することも付記する.
これまで,神経のtRNA修飾病の責任変異はヒトの疾患の研究からかなり明らかにされてきたが,発症メカニズムをtRNAや翻訳反応といった分子レベルから,細胞レベル,組織レベル,個体レベルまでつなげた研究は,ここ10年でようやく少しずつ報告されるようになった段階である.次の節では,3種類のtRNAのメチル化修飾酵素欠損マウスを用いてこれまでに明らかにされた脳のtRNA修飾病の分子メカニズムを紹介する.
5. これまでに明らかにされたtRNA修飾病の分子メカニズム
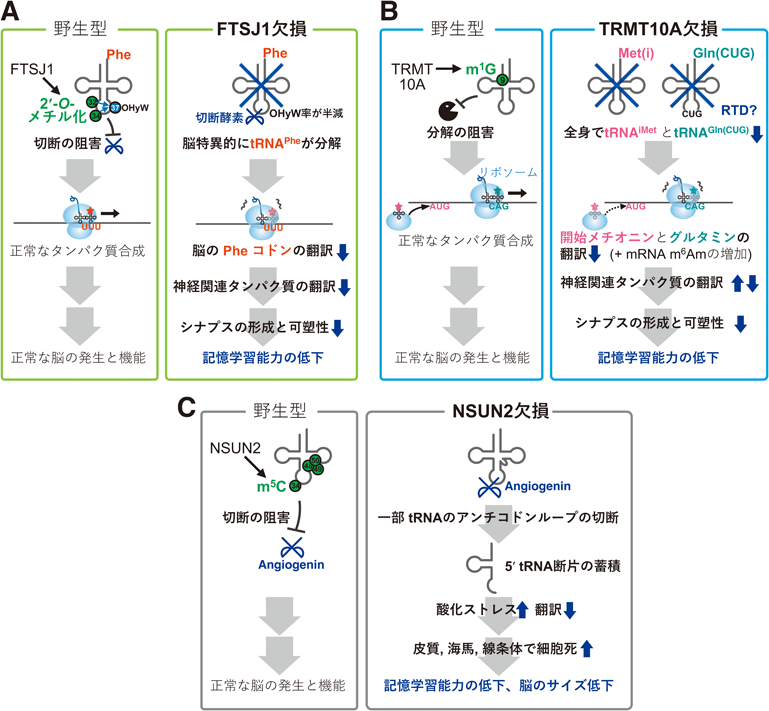
1)FTSJ1欠損による知的障害5)(図4A)
メチル化酵素FTSJ1は,さまざまな細胞質tRNAの32位と34位のリボース2′-OH基を2′-O-メチル化修飾(図1B)する.tRNAPheではさらに,32位と34位のメチル化はtRNAPhe特異的な37位のOHyW修飾の形成を促進する.また,ヒトFTSJ1遺伝子の機能欠失変異は知的障害を引き起こし,さらに自閉症スペクトラム障害を伴うこともある.このFTSJ1欠損による脳のtRNA修飾病の発症メカニズムを明らかにするために,全身Ftsj1欠損マウスが作製された5).全身Ftsj1欠損マウスでは興味深いことに,脳のみでtRNAPheのみが分解し他のさまざまなtRNAは量が変化していなかった.さらに,Ftsj1欠損マウス脳におけるtRNAPheの分解は,生後直後では緩やかで,成長に伴いtRNAPheが分解しやすくなり若い成獣ではtRNAPheが野生型よりも3割減少した.また,tRNAPheの分解は脳で起こる一方で他臓器(肝臓,腎臓)では起きない.すなわち,Ftsj1欠損のtRNAへの影響はtRNA種の特異性,組織特異性,そして時期の偏りを示していた.Ftsj1欠損脳ではtRNAPheがアンチコドンループで切られやすくなり,それに伴いtRNA全体が分解される.その結果,Ftsj1欠損脳ではmRNAのPheコドンでリボソームが止まりやすくなり,トランスクリプトームの中で特に神経関連タンパク質や膜関連タンパク質のmRNAの翻訳効率が低下していた.tRNAPhe量がほとんど減っていない幼獣脳ではPheコドンにおけるリボソームの停滞がほとんど起こらず,またtRNAPhe量がまったく減っていない肝臓ではPheコドンでのリボソームの停滞がまったく起こっていないことから,Ftsj1欠損成獣脳におけるPheコドンでのリボソームの減速は,コドン認識能の低下によるものではなくtRNAPhe量の低下によると考えられる.また,トランスクリプトームの各mRNA量がほとんど変化しなかったことから,Ftsj1欠損脳では,リボソームの停滞を介したmRNAの分解(コドンオプティマリティ)は影響していないだろう.このような神経関連mRNAの翻訳低下に伴い,Ftsj1欠損脳において細胞レベルでは,樹状突起のスパインの密度の低下と未成熟スパインの割合の増加,シナプス後肥厚の縮小がみられた.さらに,シナプス可塑性(長期増強と長期抑制)が低下し,個体レベルでは記憶学習能力の低下や不安様行動がみられた.このように,Ftsj1欠損マウスでは脳特異的なtRNAPheの分解が疾患の脳特異性を規定する可能性が示唆された.今後はさらにFtsj1欠損マウスにおけるtRNAPheの過剰発現により知的障害が緩和されるかの検証といったレスキュー実験を要するだろう.Ftsj1欠損によりtRNAPheだけが分解するというtRNA特異性の原因は,他のさまざまなtRNAは32位と34位のうちの一つまたは二つだけがFTSJ1に依存して修飾されるのに対してtRNAPheはFTSJ1に32位,34位,37位の合計三つの修飾が依存するからと考えられる.Ftsj1欠損により脳だけでtRNAPheが分解するという組織特異性と成獣になるにつれて脳でtRNAPheが分解するようになるという時期の偏りの原因は今後の解明が待たれる.
2)TRMT10A欠損による小頭症と知的障害6)(図4B)
メチル化酵素TRMT10Aは,さまざまな細胞質tRNAの9位にN1-メチルグアノシン修飾(m1G,図1B)を導入する.ヒトのTRMT10A遺伝子の機能欠失変異は,小頭症とそれに伴う知的障害を引き起こす.この変異は若年性の2型糖尿病と低身長も伴う.これらのtRNA修飾病の発症メカニズムの解明を目指してTrmt10a欠損マウスが作られた6).Trmt10a欠損マウスは患者と似てやや小さな体を持ち,脳は小頭症とまではいえない程度であるが野生型よりは若干小さく,記憶学習能力が低下していた.脳の全tRNAを定量すると,Trmt10a欠損マウスではグルタミンtRNAの一種(tRNAGln(CUG))と開始メチオニンtRNA(tRNAiMet)の計2種類のtRNAが減少していた.それに伴い,脳でグルタミンCAGコドンにおけるリボソームの減速と,開始メチオニンtRNA濃度に影響されやすいupstream open reading frameを介した翻訳開始への影響がみられた.さらに,特に神経関連タンパク質のmRNA翻訳が乱れ,シナプス後肥厚の縮小とシナプス可塑性の低下が起こり,マウス個体の記憶学習能力や協調運動能力が低下した.Trmt10a欠損マウスの脳では,Ftsj1欠損マウス脳と同様にトランスクリプトームの各mRNAの量はほとんど変化していないことから,リボソームの遅延を介したmRNAの分解はほとんど起きていないことが示唆された.
脳だけでtRNAPheが分解した全身Ftsj1欠損マウスと異なり,全身Trmt10a欠損マウスではtRNAGln(CUG)とtRNAiMetの減少が全身でみられた.一方で,Trmt10a欠損マウスの肝臓や腎臓,膵臓の機能マーカーと傷害マーカーの値は野生型と同等であった.今後はこの脳だけで組織としての明確な表現型が出る原因の究明が求められる.最近,ヒトTRMT10AはmRNAのm6A修飾の脱修飾酵素FTOのパートナーとして働きmRNAのm6A修飾の除去を助けることが報告された.Trmt10a欠損マウス脳polyA+ RNAにおいて,m6Aの全体量はほとんど変わっていなかったがm6Amは微増していた.したがって,mRNAのm6Am修飾の微増が翻訳に影響している可能性についても今後の検証を要する.
3)NSUN2欠損による知的障害7)(図4C)
メチル化酵素NSUN2は,さまざまな細胞質tRNAの34位,48位,49位,50位,およびミトコンドリアtRNAの48位,49位,50位のCを5-メチルシチジン修飾(m5C修飾,図1B)する.また,ヒトNSUN2遺伝子の変異は,小頭症,知的障害,低身長を引き起こすことが知られていた.このtRNA修飾病の発症メカニズムの解明を目的としてNsun2欠損マウスが作られた7).Nsun2欠損マウスではミトコンドリア関連の表現型はみられず,細胞質tRNAと脳への影響がみられた.具体的には,野生型マウスでNSUN2により48位や49位がメチル化される細胞質tRNAが,Nsun2欠損マウスではtRNA切断酵素Angiogeninによりアンチコドンループで切断され,約35塩基のtRNA 5′側断片が蓄積した.さらに,このtRNA断片が細胞にストレスを与え,神経細胞の翻訳量とサイズが低下し,大脳皮質,海馬,線条体で細胞死が起こり,脳のサイズ低下と記憶学習能力の低下が起こることが判明した.さらに,胎仔期に母マウスにAngiogenin阻害剤を注射することで,Nsun2ノックアウト胎仔における脳重量の減少の緩和や細胞死量の緩和がみられた.
6. 神経のtRNA修飾病のメカニズム解明と治療に向けた展望
tRNA修飾病を発症組織で大別すると,最も原因遺伝子数が多いのは,神経のtRNA修飾病である.一方で,「なぜtRNA修飾欠損は特に神経系に与える影響が大きいのか?」という問いに対する答えは,ようやく最近になり少しずつ解明されつつある状況である.全身Ftsj1欠損マウスでは,脳特異的に特定のtRNAが分解しやすいことが疾患の脳特異性を規定する一因と考えられた.一方で,全身Trmt10a欠損マウスでは全身で特定のtRNAが分解されやすい一方で組織としての機能低下は脳だけで顕著にみられた.興味深いことに,Ftsj1欠損マウス,Trmt10a欠損マウス,Nsun2欠損マウスのすべてで共通していたことは,tRNA修飾を失うと限られた種類のtRNAが切断,分解されることである.したがって,切断・分解酵素を適切なタイミングで阻害すれば,このようなメカニズムで発症する神経のtRNA修飾病を緩和できるかもしれない.あるいは,tRNA修飾欠損によりどのtRNAの量が減少するかが明らかになれば,そのtRNAの発現ベクター(アデノ随伴ウイルス等)を神経系に導入して当該tRNAを過剰発現させれば,FTSJ1やTRMT10AのようにtRNA量の不足が大きな原因と考えられる疾患の緩和に有用かもしれない.コドン認識に直接関与するtRNAアンチコドン1文字目の修飾欠損についても,酵母や線虫の実験系において,対応するコドンの翻訳スピードの低下がタンパク質凝集の原因であることと,そのtRNAの過剰発現により翻訳スピードとタンパク質のフォールディングが回復することが示されている.したがって,各修飾の欠損によりどのtRNAがクリティカルに影響を受けるかを明らかにすれば,多くの修飾については,tRNAの過剰発現で病態が改善する可能性が考えられ,今後のマウス等を使った検証が重要になるだろう.
本稿で紹介した三つのマウスの研究は,tRNA修飾病の発症メカニズムの一端を解明したという点で先駆的である.一方,いずれの研究もtRNA修飾欠損というtRNA分子の変化から脳の表現型までを何とか一本の細い道でつないだにすぎず,発症メカニズムをより広く,深く,細かく照らし出すことが必要である.脳は神経細胞とグリア細胞(アストロサイト,オリゴデンドロサイト,ミクログリアなど)などから構成されるが,これまでのマウスを用いた研究におけるtRNA-seqやリボソームプロファイリング実験は全脳のさまざまな細胞の平均をみている.今後は細胞種特異的ノックアウトマウスや細胞種特異的リボソームプルダウン技術,イメージング技術などを使うことで細胞種ごと,脳の部位ごとの解析も必要だろう.
神経組織における翻訳の特殊な点として,細胞体から遠く離れた樹状突起や軸索においても翻訳(局所翻訳)が起こり,しかも高頻度なシナプス伝達が起こるとその近傍で局所翻訳が活性化されることがあげられる.また,局所翻訳されたリボソームタンパク質が神経突起内のリボソームに結合するという通常の細胞では考えられないような現象も報告されている.局所翻訳は,シナプスの可塑性とそれによる長期記憶に必須であり,シナプスに対する刺激前後において,tRNA修飾の有無が局所翻訳にどう影響するかも今後の検証が待たれる.
mRNAは顆粒状の構造体の形で神経突起内を輸送される.一方,軸索や樹状突起におけるtRNAの動態はほとんどわかっていないのが現状である.したがって,tRNA修飾がtRNAの神経突起におけるtRNAの局在や機能にどう影響しているかもほぼ未解明である.たとえば,特定のtRNA修飾酵素の欠損により特定のtRNAが分解する場合,その修飾欠損tRNAは転写された核から時には数十センチという距離を輸送され,シナプス近傍に到達するころには細胞体における濃度よりさらに少なくなり,局所翻訳の効率が著しく低下するような例はあるのだろうか? 神経でtRNA修飾病が最も多くみられるという事実は,神経におけるtRNA修飾の大切さを端的に示しており,ここに多くの重要な研究テーマが埋まっているだろう.
引用文献References
1) Suzuki, T. (2021) The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol., 22, 375–392.
2) Chujo, T. & Tomizawa, K. (2021) Human transfer RNA modopathies: diseases caused by aberrations in transfer RNA modifications. FEBS J., 288, 7096–7122.
3) Asano, K., Suzuki, T., Saito, A., Wei, F.-Y., Ikeuchi, Y., Numata, T., Tanaka, R., Yamane, Y., Yamamoto, T., Goto, T., et al. (2018) Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res., 46, 1565–1583.
4) Wei, F.-Y., Suzuki, T., Watanabe, S., Kimura, S., Kaitsuka, T., Fujimura, A., Matsui, H., Atta, M., Michiue, H., Fontecave, M., et al. (2011) Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Invest., 121, 3598–3608.
5) Nagayoshi, Y., Chujo, T., Hirata, S., Nakatsuka, H., Chen, C.-W., Takakura, M., Miyauchi, K., Ikeuchi, Y., Carlyle, B.-C., Kitchen, R.-R., et al. (2021) Loss of Ftsj1 perturbs codon-specific translation efficiency in the brain and is associated with X-linked intellectual disability. Sci. Adv., 7, eabf3072.
6) Tresky, R., Miyamoto, Y., Nagayoshi, Y., Yabuki, Y., Araki, K., Takahashi, Y., Komohara, Y., Ge, H., Nishiguchi, K., Fukuda, T., et al. (2024) TRMT10A dysfunction perturbs codon translation of initiator methionine and glutamine and impairs brain functions in mice. Nucl. Acids Res., 52, 9230–9246.
7) Blanco, S., Dietmann, S., Flores, J.-V., Hussain, S., Kutter, C., Humphreys, P., Lukk, M., Lombard, P., Treps, L., Popis, M., et al. (2014) Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J., 33, 2020–2039.
著者紹介Author Profile
 中條 岳志(ちゅうじょう たけし)
中條 岳志(ちゅうじょう たけし)熊本大学大学院生命科学研究部(医)講師.博士(工学).
略歴2007年東京大学工学部卒業.12年同大学院修了,博士(工学).北海道大学博士研究員を経て,18年熊本大学大学院生命科学研究部(医)富澤一仁研究室の特任助教,22年より現職.
研究テーマと抱負tRNA修飾病の分子機構の解明と人為的なRNA修飾の導入技術の開発.
ウェブサイトhttps://researchmap.jp/TakeshiChujo
趣味オオクワガタの飼育.