小児期発症の胆汁うっ滞性肝疾患(pediatric cholestatic liver disease:Ped-CLD)は超希少疾患の集合体である.新生児のうち2500人に1人の頻度で発症するが,その原因,臨床経過は多様であり,治療法のない難治性の疾患も多数含まれる.本疾患は,臨床所見,遺伝子検査,肝病理検査等の統合的な解釈をもって確定診断が行われているが,うち75%は原因の特定に至らない.
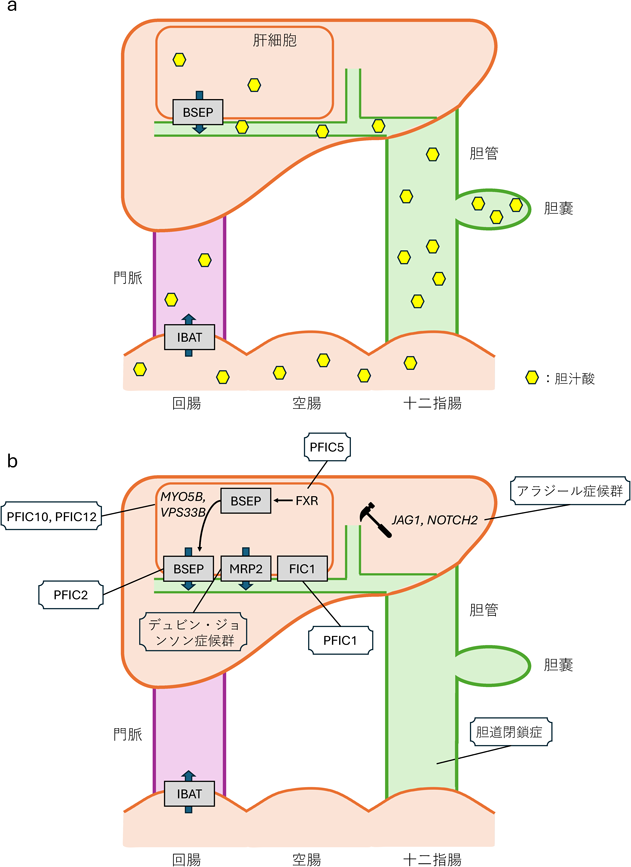
胆汁は,肝臓からの異物排泄,小腸における脂質の乳化と吸収促進など,さまざまな機能を担う液体である.主成分は水や無機物質で血漿と等浸透圧だが,胆汁酸,リン脂質,コレステロールをはじめとした有機化合物も含んでいる.胆汁は肝細胞から毛細胆管へ分泌されると肝内胆管を経て肝管へと集約され,胆嚢で濃縮を受けたのちに総胆管から小腸へ排泄される.毛細胆管への有機化合物の排出は能動輸送であり,肝実質細胞の毛細胆管側膜に発現するbile salt export pump(BSEP)をはじめとするABCトランスポーターがその機能を担う.BSEPは胆汁酸を基質とする.BSEPを介した毛細胆管中への胆汁酸排泄は,浸透圧勾配を形成し,胆汁流形成の主要な駆動力となる.なお,胆汁排泄された胆汁酸のほとんどは回腸で再吸収を受け,門脈を介して肝臓へと戻る(腸肝循環,図1a).
胆汁うっ滞とは,胆汁生成および流出の過程において,何らかの原因により胆汁の流れが滞り,結果として肝臓内への胆汁成分の蓄積や血液中への流出,ひいては黄疸および肝障害を引き起こす病態である.Ped-CLDでは,BSEPをコードするABCB11の遺伝子異常により発症する進行性家族性肝内胆汁うっ滞症2型(progressive familial intrahepatic cholestasis type 2:PFIC2)のように,胆汁うっ滞の成因が明確な場合もあるが1),その多くでは成因不明である.胆汁うっ滞を呈する疾患としては,Ped-CLD以外にも多岐にわたり,アルコール性肝障害,薬物性肝障害,原発性硬化性胆管炎,急性肝炎,胆管結石などが知られている.
Ped-CLDは,その発症機序について理解が進んでいないため,本疾患全体を対象とする内科的治療法は確立していない.肝障害が進行して肝硬変へと進行した場合などは,肝移植の対象となる.肝移植により根治する患者がいる一方で,自己抗体の産生や脂肪肝炎の発症により,ドナー肝が機能しない(グラフト不全)患者も存在する.また肝移植にはドナー不足の問題や,生涯にわたる免疫抑制剤の服用等の課題も残されている.したがって,Ped-CLDの病態発症,進展を規定する機序を解明し,それに応じた内科的治療法を開発することは喫緊の課題である.
代表的なPed-CLD(図1b)について胆汁うっ滞の発症機構を以下に記す.
1)胆道閉鎖症2)
胆道閉鎖症は,胆汁流出経路の閉塞により生じる疾患で,発症率が1万出生につき1人程度の希少疾患である.生後まもなく遷移性黄疸,便色異常,濃黄色尿によって顕在化する.閉塞部位に応じて型に分類され,I型は総胆管,II型は肝内胆管,III型は肝門脈が閉塞されている.原因は先天性,ウイルス感染,母体要因などさまざまな可能性があげられているが,未解明である.そのため治療法は外科的治療に限られる.まず閉塞部位を切除し肝管腸吻合術や肝門部腸吻合術により胆道再建と胆汁流出を図るが,奏功しない場合は肝移植を行う.
2)進行性家族性肝内胆汁うっ滞症(PFIC)1)
PFICは常染色体潜性遺伝疾患で,発症率は5万~10万出生につき1人程度の超希少疾患である.生後まもなく遷移性黄疸の出現によって顕在化し,直接ビリルビン優位の黄疸や,AST, ALT, 総胆汁酸高値などの胆汁うっ滞所見を示す一方でγ-GTP値が不相応に低いことが臨床的特徴である.
原因遺伝子に応じて執筆時点では12の型(PFIC1~12)に分類される.2型の原因遺伝子であるABCB11は,前述のように肝細胞から毛細胆管中への胆汁酸排泄を担うBSEPをコードしている.そのためPFIC2患者ではBSEPが機能異常を来し,肝内胆汁うっ滞が生じる.PFICの原因遺伝子には,BSEPの転写(NR1H4; PFIC5),細胞膜移行(MYO5B; PFIC10, VPS33B; PFIC12),胆汁酸輸送活性(ATP8B1; PFIC1)の制御に働く遺伝子も含まれている.これら遺伝子異常は,BSEPの機能低下により,胆汁うっ滞を惹起すると考えられている.その他の原因遺伝子としては,毛細胆管の形成維持に働く遺伝子が複数同定されている.これらの遺伝子異常時には,毛細胆管内に胆汁酸を濃縮できないため,胆汁流が低下し,病態が発症すると推察されている.
3)アラジール症候群3)
アラジール(Alagille)症候群は常染色体顕性遺伝疾患で,有病率はおよそ3万出生に1人の超希少疾患である.本疾患の原因遺伝子はJAG1, NOTCH2である.小葉間胆管の低形成が肝組織学的な特徴であり,本形態異常が胆汁流の低下,ひいては胆汁うっ滞発症につながる.臨床的な特徴は,特徴的顔貌・眼球異常・先天性心奇形・椎骨奇形などの肝外症状である.JAG1, NOTCH2ともに発生段階においてNotchシグナルを介した細胞分化の制御に関わるため,多臓器にわたり病態が発症すると考えられている.
4)その他の疾患
デュビン・ジョンソン(Dubin-Johnson)症候群は,毛細胆管側膜で直接ビリルビン輸送を担うmulti-drug resistance protein 2(MRP2)をコードするABCC2の変異により発症する.MRP2の機能異常の結果,直接ビリルビンの肝細胞での蓄積,ひいては高ビリルビン血症をもたらす.また,シトリン欠損による新生児肝内胆汁うっ滞症(neonatal intrahepatic cholestasis caused by citrin deficiency:NICCD)は,輸送タンパク質シトリンをコードするSLC25A13の変異により発症するが,本遺伝子異常により胆汁うっ滞が発症する機序は不明である.このように原因遺伝子は判明しているものの病態発症機構が不明な疾患は,NICCD以外にも多数知られている.
1)フェニル酪酸ナトリウム(NaPB)
筆者らは,胆汁流形成の基点となるBSEP機能を増強できれば,Ped-CLDの治療法開発につながるのではないかと考えた.in vitroスクリーニングの結果,尿素サイクル異常症の治療薬であるNaPBのBSEP発現量増強作用(新規薬理作用)を発見した4).非臨床データを取得後,本薬剤のPFIC患者に対する有効性・安全性の探索を目的とした臨床研究を実施した.NaPBの投与により肝病理所見,臨床所見(黄疸,掻痒)が改善した5).自己肝生存期間の著明な延長を示唆する結果も得られた6).本効能について薬事承認を取得すべく研究者主導での治験を実施し,臨床研究から想定された結果を得るに至っている.
2)回腸胆汁酸トランスポーター(IBAT)阻害薬
胆汁うっ滞に伴う掻痒感は,患者および患者家族のquality of life(QOL)を著しく低下させる臨床症状である.睡眠障害をもたらすことも多く,掻痒感がきわめて強い場合は自殺念慮を抱く患者も存在する7).皮膚に病変はなく,痒みをもたらす原因については,胆汁うっ滞に伴い胆汁酸の血液中濃度が上昇することを基盤としてさまざまな仮説が古くから提唱されている.一方,近年ではオートタキシン(ATX)の活性化によるリゾホスファチジン酸(LPA)の増加も痒みの原因としてあげられている.胆汁排泄を受ける物質にATX活性化効果を持つものが含まれると推測されている7).
回腸胆汁酸トランスポーター(ileal bile acid transporter:IBAT)阻害薬は,回腸における胆汁酸再吸収を阻害し,胆汁酸の腸肝循環を抑制することで体内の胆汁酸プールを減弱させる作用を持つ.欧米諸国では,Ped-CLDのうちアラジール症候群・PFIC(米国のみ)による掻痒症を適応として承認がなされている.本邦でも,本年に武田製薬より製造販売承認申請がなされたところである.
4. Ped-CLDのさらなる理解と治療法開発に向けた取り組み
Ped-CLDは多くの超希少疾患から構成されるため,疾患理解に資するリソースが国内外を問わず集約できていない.またヒトの病態を正確に反映する適切な動物モデルが存在しない.これらは,Ped-CLDの病態理解,治療法開発における障壁となっている.本課題解決に向け,以下の取り組みがなされている.
1)患者登録研究(CIRCLe)
Ped-CLDの研究基盤構築を目的とし,筆者らは患者登録研究CIRCLeを2021年に開始した.本研究は,Ped-CLDの診断・診療支援(遺伝子検査,胆汁酸分析,肝病理評価など)が登録の入口となっている.登録後は定期的な追跡調査を実施し,登録症例の診療情報,生体試料を前向きに収集している.執筆時点では,共同研究機関として38都道府県・92医療機関が参画している.臨床情報を糸口とした病態解明研究により,PFIC1において肝移植後グラフト不全が生じる原因8)を特定する等,患者リソース集約による成果が芽吹きつつある.
2)動物モデルの作出
患者の病態を再現する動物モデルを構築すべく,実験系が模索されている.以下に,Ped-CLDの研究で汎用されている実験動物モデルを紹介する.
a.マウス
マウスはヒトと胆汁酸組成が異なる.げっ歯類では細胞障害性が低い親水性胆汁酸が多いのに対して,ヒトでは,細胞障害性が高い疎水性胆汁酸が多い.Ped-CLDの原因遺伝子をマウスでノックアウト(KO)した際に患者の病態を再現できない理由は,この種差に起因すると認識されている.そこで,両種間における胆汁酸組成の隔たりを解消するために以下の手法が考案されている.
①コール酸添加食の給餌による負荷
従来用いられてきた簡便な手法が,胆汁酸の一種であるコール酸を付加した餌を与えることで,ヒトの胆汁酸組成へ外因的に近づけるものである.
Wangらは,PFIC2の原因遺伝子であるAbcb11 KOマウスは通常軽度の胆汁うっ滞しか呈さないのに対し,コール酸を付加した餌を与えると,黄疸,肝臓の壊死などをはじめとした,患児で認められる重篤な胆汁うっ滞所見を示すことを発見した9).またHanleyらは,ARC症候群およびPFIC12の原因遺伝子であるVps33bを肝細胞・胆管細胞特異的にKOしたマウスで確認される血液生化学,肝組織学の軽微な異常が,コール酸負荷により患者病態と類似した重篤な表現型となることを報告している10).
②げっ歯類特異的な胆汁酸代謝酵素の欠損
マウスをはじめとしたげっ歯類とヒトとの間で認められる胆汁酸組成の種差は,げっ歯類特有の胆汁酸代謝酵素Cyp2c70, Cyp2a12の機能に起因する11).Cyp2c70は,ヒトも有するケノデオキシコール酸(CDCA)をげっ歯類特有のミュリコール酸(MCA)に代謝する.CDCAは疎水性で細胞毒性がある一方,MCAは親水性で細胞保護作用がある.Cyp2a12は,腸内細菌により脱水酸化された二次胆汁酸を一次胆汁酸であるコール酸(CA)とCDCAに再水酸化する働きがある.本田らは,両酵素をKOしたマウスを作出し,MCAの消失,ならびに胆汁酸組成が疎水化することを確認している11).
筆者らは,肝臓特異的にCas9タンパク質を発現するトランスジェニックマウスに,Cyp2c70に対するsingle guide RNA(sgRNA)を導入することにより,後天的にマウスの胆汁酸組成をヒト型に変換する実験系を構築した12).sgRNAの導入には,肝臓指向性の高いアデノ随伴ウイルスベクターを用いた.本実験系で,Abcb11とCyp2c70を併せてノックダウン(KD)すると,Abcb11単独でのKD時に比べ,患児に近い肝病理像を取得ができた12).現在,Abcb11以外の胆汁うっ滞関連遺伝子についても本実験系にて解析を進めている.
b.ゼブラフィッシュ
ゼブラフィッシュは,受精卵に対するインジェクションが可能であり,発生が早く3日齢程度で孵化するため,発生段階の遺伝子機能解析に資する動物モデルとして汎用されている.また幼生は体が透明なため,蛍光タンパク質発現遺伝子の導入や蛍光物質の投与により,臓器形成や胆汁蓄積・流出の様子を可視化し,観察することができる.
Lorentらは,ゼブラフィッシュにおいてアラジール症候群の原因となるJAG1, NOTCH2のオーソログをそれぞれあるいは複合的にKOすると,患児と同様に肝内胆管発達の顕著な異常を呈することを確認した13).本研究を通じ,ゼブラフィッシュが胆管研究のモデルに資すること,Jagged遺伝子を介したNotchシグナルが肝芽細胞の運命決定に寄与している可能性が明らかとなった.Ellisらは,PFIC2の原因遺伝子ABCB11のオーソログであるAbcb11bをKOすると,胆汁酸の胆汁排泄が滞り,細胞の膨張や壊死,毛細胆管の拡張と微絨毛の消失など,患児の臨床所見と類似した表現型を呈することを報告している14).また,Karolczakらは,X鎖連鎖性ミオチューブラーミオパチー(X-linked myotubular myopathy:XLMTM)の原因遺伝子Mtm1をゼブラフィッシュにおいて欠損させ,マウスでは確認できなかった患児の肝病態(胆汁うっ滞など)の再現に成功した15).エンドソームの輸送制御を担うMTM1の分子機能と整合性のある病態発症モデルを提唱するに至っている.