1)肺がんの罹患数と生存率
世界では,毎年2000万人以上ががんに罹患し,970万人ががんで亡くなっているとされる.中でも肺がんはがんによる死亡の主な要因の一つであり,2022年には約180万人が肺がんで亡くなり,これは全がん死亡数の約20%に相当する.わが国では年間40万人弱ががんで亡くなり,全死亡の25%を占める.肺がんによる死亡者数は76,600人で,そのうち約7割が男性である.5年相対生存率は35%と20年前に比べると大幅に改善してきたものの,完治が困難ながん種である.
肺がんの死亡者数が多い理由として,発症数が年々増加し,新規症例が2022年には250万件に達したこと(男性では前立腺がん,大腸がんに次いで3番目に多く,女性では大腸がん,乳がんに次いで3番目に多い)や,初期で発見されることが比較的少なく,進行した状態で発見されることがあげられる.肺がんの原因としては喫煙や大気汚染などが考えられるが,加齢に伴う遺伝子変異の蓄積も発症要因とされている.特に喫煙は,肺がん全体の3~4割を占める特定のタイプに関連し,そのタイプでは患者の95%以上が喫煙者であることから,禁煙を推進することで患者数を約2/3に減少させることが可能であると推定される.
肺は生存に必須の臓器であり,手術で切除できる範囲が限られるため,肺がんが進行し複数の病変が出現すると,手術での根治的切除が不可能となる.そのため,予防と早期発見が非常に重要であり,世界的には低線量CT検査などを用いたスクリーニングが早期治療の鍵とされている.しかし,わが国では低線量CTが公費検診の助成対象となっておらず,健康診断時には胸部単純X線検査が広く行われている.この検査では検出可能な肺がんのサイズが比較的大きい(1 cm前後)ため,課題とされている.
アメリカ国立がん研究所(NCI)の大規模臨床試験では,50~74歳のヘビースモーカーを対象に低線量CT検診を行った結果,単純X線検診より肺がん死亡率が20%減少することが報告されている1).この結果を受け,オランダやベルギーでも同様の報告があり,一定年齢以上の喫煙者には低線量CT検診が推奨されている.一方で,非喫煙者や軽喫煙者での効果については定まった結論が出ておらず,今後の研究が待たれる.
2)肺がんのタイプ
肺がんは,病理組織学的に分類され,大きく二つに分けられる.非小細胞肺がんと小細胞肺がんである.非小細胞肺がんは主に,肺腺がん,肺扁平上皮がん,それ以外に分類される.
この中で肺腺がんは,非小細胞肺がんの中で最も一般的であり,肺がん全体の約50%を占める.女性や非喫煙者にも多くみられ,主に肺の外側部分に発生するのが特徴である.肺腺がんは肺の分泌腺を構成する細胞から発生すると考えられており,小細胞肺がんなどと比べて増殖が比較的遅いものの,従来から用いられてきた殺細胞性の抗がん剤に対しては,やや抵抗性を示すことが多いとされている.肺腺がんでは,ドライバーがん遺伝子と呼ばれる遺伝子異常が原因となってがんが発生する場合が多く,その遺伝子異常を標的とした分子標的薬が,この20年間で多数開発され,目覚ましい治療効果を上げてきた.
細胞の成長や増殖に直接関与する遺伝子であり,この遺伝子に変異が生じることで,過剰な増殖シグナルが恒常的に活性化され,細胞が無秩序に増殖し,がんが発症する.肺がんにおける代表的なドライバーがん遺伝子の変異には,以下のものがある.
1)EGFR遺伝子変異
EGFR(上皮成長因子受容体)遺伝子の変異は,特に非小細胞肺がん患者に多くみられる.EGFRは通常,細胞膜に発現する1回膜貫通型の受容体型チロシンキナーゼである.細胞外の成長因子(EGFなどのリガンド)が,EGFRの細胞外領域に結合すると,構造変化を起こし,細胞内のチロシンキナーゼ領域が活性化し,細胞の生存・増殖シグナルを細胞内へと伝えていく.しかし,EGFR遺伝子に変異が生じると(主に細胞内のチロシンキナーゼ領域内に1アミノ酸の置換または,5アミノ酸の欠失変異が生じる),成長因子がなくてもEGFR受容体が常に活性化し,細胞内のチロシンキナーゼが常に活性化した状態となり,細胞増殖シグナルが持続的かつ過剰に伝えられる2).その結果,細胞は制御不能な増殖を続け,がん細胞として増殖を続ける.EGFRの変異は,日本人肺腺がんの約半数に見つかるが,世界的には肺腺がんの約15%程度とされる3).
2)KRAS遺伝子変異
KRASは,細胞の成長と分裂を制御するMAPK(mitogen-activated protein kinase)シグナル伝達経路に関与する遺伝子である.KRASは,GTP(グアノシン三リン酸)結合型の活性型とGDP(グアノシン二リン酸)結合型の不活性型をGAP(GTPase活性化タンパク質)およびGEF(グアニンヌクレオチド交換因子)活性を担うタンパク質が切り替えることで,RAFタンパク質と結合し,下流のMAPK経路の活性化を促進する.RAFにはARAF, BRAF, CRAFの三つのファミリータンパク質があるが,活性化したKRASは主にCRAFやBRAFとの結合を介して,さらに下流のMEK, ERKの活性化を誘導する.がんにおけるKRASの変異は,塩基置換により主に12番目および13番目のグリシン(G)がほかのアミノ酸に置き換わるものである.これによりKRASのGTP加水分解活性が低下し,KRASが常にGTP結合状態を維持してしまう.すると,GTP結合型KRASに結合する下流分子が恒常的に結合できるようになり,RAS/RAF/MEK/ERKやPI3K/AKTなどのシグナル伝達経路が恒常的に活性化される.他のKRAS変異としてはQ61の変異やA146T変異などが見つかっているが,大半はG12またはG13の変異であり,G12がシステインに代わるG12Cに対しては,変異したシステイン残基に共有結合し特異的に阻害できる薬剤が複数開発され,一部はKRAS陽性肺がんの治療法の一つとなっている.KRASの変異は,喫煙者の非小細胞肺がん症例(特に肺腺がん)に比較的多い傾向がある4).
3)ALK融合遺伝子
ALK(未分化リンパ腫キナーゼ)は,未分化大細胞非ホジキンリンパ腫(ALCL)で発見された受容体型チロシンキナーゼであり,リガンドとしてはALK-ALが近年同定されている.2007年には肺がんにおいてEML4–ALK(echinoderm microtubule associated protein like-4–anaplastic lymphoma kinase)融合遺伝子が発見され,その後,さまざまな固形がんでもALK融合遺伝子が確認されている5, 6).ALK融合遺伝子は,ALKのチロシンキナーゼにあたるC末端側半分をコードする遺伝子領域と,二量体・多量体化能を持つ細胞骨格タンパク質などをコードする遺伝子が,染色体の転座や逆位により形成される.通常ALKは成人では脳のごく一部の細胞でのみ発現し,肺組織ではほとんど発現しない.しかし,融合遺伝子を形成することで融合相手の転写活性が活性化され,ALK融合タンパク質が発現し,がん化を誘導する.さらに,ALKはリガンドによる制御を受けるが,多量体化能を持つパートナー分子と融合遺伝子を形成することで,ALK融合タンパク質は二量体・多量体を形成し,ALKチロシンキナーゼが恒常的に活性化する.この結果,ALK融合タンパク質は自身のC末端領域のチロシン残基やほかの結合タンパク質のチロシン残基をリン酸化し,RAS/RAF/MEK/ERKやPI3K/AKTなどの下流シグナル経路を恒常的に活性化し,がん化を促すとされる.ALK陽性肺がんの特徴として,若年層や非喫煙者の肺腺がん患者に多くみられることがあげられる.ALK融合遺伝子のみで強力に細胞をがん化させる能力を持つと考えられ,がん抑制遺伝子の機能欠失変異などを伴わない場合も多いとされる.そのため,ALK融合遺伝子陽性のがん細胞はALKからの増殖シグナルに強く依存しており,ALK阻害薬によって顕著かつ持続的な抗腫瘍効果が認められている.
4)ROS1融合遺伝子
ROS1遺伝子もALKと同様に受容体型チロシンキナーゼをコードしているが,そのリガンドはごく最近まで不明であった.近年,ROS1の内在性リガンドとしてNELL2(neural EGFL like 2)が同定された7).ROS1の細胞内チロシンキナーゼ領域は,ALKと非常に高い相同性を持ち,アミノ酸配列の70%以上が一致している.ROS1融合遺伝子は,肺がんの約1%程度で確認されるほか,胆道がんや膵臓がんの一部でも見つかっている8–11).ALKと同様に,ROS1融合遺伝子も融合パートナー遺伝子(CD74やSLC34A2など)によって二量体・多量体化した融合タンパク質を形成することで,ROS1チロシンキナーゼが恒常的に活性化し,MAPKやPI3Kなどの細胞増殖シグナルを恒常的に活性化する.さらに,ROS1のキナーゼ領域がALKと高い相同性を持つことから,一部のALK阻害薬がROS1阻害活性も有しており,ROS1融合遺伝子陽性肺がんの治療薬として用いられている.
5)BRAF活性化変異
BRAFはセリントレオニンキナーゼタンパク質であり,その活性化変異は皮膚がん(メラノーマ)や一部の甲状腺がんで見つかり,その多くはV600E変異を有する12).肺がんにおけるBRAF活性化変異は,非小細胞肺がんの約1~3%で見つかる13).この変異は,MAPK経路(RAF/MEK/ERK経路)を恒常的に活性化させ,がん細胞の増殖シグナルを恒常的に活性化する.そのうちV600E変異は最も高頻度で認められるBRAF変異で,特に肺がんと悪性黒色腫では頻繁にみられる.V600E変異はBRAFの構造を変化させ,GTP型RASの存在に関係なく常に活性化状態を保つ.V600E以外のBRAF変異として,たとえばG469AやT599Iなどが報告されているものの,変異の種類によって,その活性化様式が異なる14, 15).これらの変異体はV600Eほどの強い活性化は示さず,V600E特異的な阻害剤にはほとんど影響を受けない.V600E変異型BRAFには特異的阻害薬が複数開発され臨床応用されているが,非V600E変異の場合,現在臨床で用いることのできる阻害薬はない.今後,非V600Eにも阻害活性を示す薬剤が開発され臨床応用されることを期待したい.
6)RET融合遺伝子
RET融合遺伝子は,RET(rearranged during transfection)遺伝子がほかの遺伝子と融合することで形成されるがん遺伝子であり,肺腺がんの1~2%で見つかる11, 16).ほかには甲状腺がんや大腸がんのごく一部でも低頻度ながら見つかる.RETは受容体型チロシンキナーゼをコードする遺伝子で,細胞の成長や分化の調整に関与する.そのリガンドとしては,GDNF(glial cell line-derived neurotrophic factor)ファミリーの成長因子が知られており,主に,GDNF, NTRN(Neurturin), ARTN(Artemin), PSPN(Persephin)などがある.その活性化メカニズムは特徴的であり,リガンドは直接RETに結合するのではなく,まずGFRα(GDNF Family Receptor α)と呼ばれる補助受容体と結合する(GDNFにはGFRα1, NTRNにはGFRα2).次に,その複合体がRETの細胞外領域に結合し,RET受容体が二量体化することで,RETのチロシンキナーゼ領域が活性化され,下流のシグナル伝達経路(MAPK経路,PI3K/AKT経路など)が活性化される.融合遺伝子を形成した場合には,ALK同様に,融合パートナー[KIF5B(kinesin family member 5B)やCCDC6(coiled-coil domain containing 6)など]によって二量体化・多量体化され,RETチロシンキナーゼの活性化が生じる.そのため,RET融合遺伝子陽性肺がんには,RET選択的なチロシンキナーゼ阻害薬(セルペルカチニブなど)が高い選択的阻害活性を示し,現在これらの阻害薬が承認され臨床応用されている17).
7)その他
EGFRファミリーに属するHer2(ERBB2)の活性化変異(Exon20に1~数アミノ酸の挿入変異),NTRK(neutrophic tropomyosin receptor kinase)融合遺伝子(チロシンキナーゼであるNTRK1, NTRK2またはNTRK3が融合遺伝子を形成)やLTK(leukocyte receptor tyrosine kinase)融合遺伝子18)が見つかり,これらはいずれも相互排他的(同一症例ではほとんどこれらを二つ以上持つケースは見つからない)である.
また,小細胞肺がんでは,これら融合遺伝子などのドライバーがん遺伝子変異が見つかることはほとんどなく,TP53遺伝子変異とRb遺伝子の機能喪失変異が高頻度に見つかるが,直接的な治療標的となるドライバーがん遺伝子は報告されていない.なお,TP53は,がん抑制遺伝子の一つで,細胞が異常な増殖を起こしたときにその増殖を止めたり,細胞死を促したりする役割を持つ.しかし,TP53遺伝子に変異が生じると,その抑制機能が失われ,異常な細胞が生き残りやすくなる.このTP53の変異は,約半数以上のがんで認められ,がんで最も高頻度に認められる遺伝子変異である.他に,非小細胞肺がんの15%程度を占める扁平上皮肺がんでは,FGFR1の遺伝子増幅が~10%の症例で見つかるとされるものの,治療標的にもなりうる,いわゆるドライバーがん遺伝子の報告はほとんどない[PIK3CA(phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α)の変異も認められるものの,その阻害だけでは十分な抗腫瘍効果が得られず,他の因子と併せて阻害することが必要と考えられている]19).
肺がんの治療には,上述したような特定の遺伝子変異やタンパク質の異常を標的にして,がん細胞の増殖を抑制する「分子標的薬」がこの20年の間に広く利用されるようになった.特にEGFR, HER2, ALK, ROS1, RET, BRAF, NTRK, KRAS(今はG12Cのみ承認薬がありほかのKRAS変異阻害薬は多数開発中)などの遺伝子変異や融合遺伝子が存在する場合,これらに対応した薬剤が高い有効性を示す.次に,主要な分子標的薬の種類と対象となる遺伝子変異について概説する(表1).
表1 肺がんドライバーがん遺伝子とその変異様式,並びに分子標的薬法| ドライバー遺伝子 | 変異様式(分子性状への影響) | 分子標的薬 |
|---|
| EGFR遺伝子変異 | 細胞内チロシンキナーゼ内変異(受容体が恒常的活性化) | (第一世代)ゲフィチニブ,エルロチニブ
(第二世代)アファチニブ,ダコミチニブ
(第三世代)オシメルチニブ
(抗EGFR–MET抗体)アミバンタマブ |
| KRAS遺伝子変異 | G12C変異(GTP結合状態を維持し恒常的活性化) | ソトラシブ,アダグラシブ(本邦未承認) |
| ALK融合遺伝子 | パートナー分子(EML4)との融合遺伝子形成(多量体形成による恒常的活性化) | (第一世代)クリゾチニブ
(第二世代)アレクチニブ,セリチニブ,ブリグチニブ
(第三世代)ロルラチニブ |
| ROS1融合遺伝子 | パートナー分子(CD74, SLC34A2)との融合遺伝子形成(多量体形成による恒常的活性化) | クリゾチニブ,エヌトレクチニブ,レポトレクチニブ |
| BRAF活性化変異 | V600E変異(BRAFの構造変化,恒常的活性化)
非V600E変異(変異により異なる) | ダブラフェニブ,ベムラフェニブ,エンコラフェニブ |
| RET融合遺伝子 | パートナー分子(KIF5B, CCDC6)との融合遺伝子形成(多量体形成による恒常的活性化) | セルペルカチニブ,プラルセチニブ(本邦未承認) |
| その他 | ・Her2変異(恒常的活性化)
・NTRK融合遺伝子形成
・LTK融合遺伝子形成 | (HER2抗体薬物複合体)トラスツズマブデルクステカン
(NTRK阻害薬)ラロトレクチニブ,エヌトレクチニブ
(MET阻害薬)カプマチニブ,テポチニブ,グマロンチニブ |
1)EGFR阻害薬
EGFRの活性化変異は約45%がExon21のL858R変異,約40%がExon19の約5アミノ酸欠損(Del19)からなるが,その他にもExon20に1~数アミノ酸が挿入する変異などさまざまなパターンの活性化変異が発見されており,下記のEGFRチロシンキナーゼ阻害薬(EGFR-TKI)が使用されている.
a.第一世代:ゲフィチニブ,エルロチニブ
EGFRの最も高頻度に認められる変異(L858R変異,Del19変異)などを持つ患者に使用され,EGFR阻害薬として広く使われてきた.
b.第二世代:アファチニブ,ダコミチニブ
効果が第一世代より強力(低濃度でEGFRを阻害できる)で,第一世代と異なりEGFRの797番目のシステイン残基(C797)に共有結合することで強力な阻害活性を持つ.Exon20挿入変異を除くマイナーなEGFR活性化変異にも広く阻害活性を有する.
c.第三世代:オシメルチニブ
第一,第二世代EGFR-TKIに耐性を示すEGFR-T790M耐性変異体にも高い阻害活性を有しており,第一,第二世代のEGFR-TKI治療後に耐性が生じた場合にも使用できる20).もちろん,初回治療として使用しても高い抗腫瘍効果が得られており,第一,第二世代EGFR-TKIよりも長い無増悪生存期間(progression-free-survival:PFS)が示されている21).また,脳への移行がよく,脳転移にも効果を示すことが確認されている.
d.抗EGFR–MET二重特異的抗体
EGFR-Exon20挿入型変異には,上記のEGFR阻害薬の効果は乏しく,臨床的有用性はほとんど認められない.一方で,近年になり,一つの抗体分子に二つ異なる抗原認識部位を持たせることで,二つの抗原を同時に標的とできる人工的な抗体が開発された.特にEGFRとMETを一つの抗体で認識できる抗体(Amivantamab)がEGFR-Exon20陽性肺がん患者において高い抗腫瘍効果を認めたため,承認され臨床応用されている22, 23).
2)KRAS阻害薬
KRASの活性化変異で最もメジャーなものは前述のとおり12番目のグリシン(G)が変異するものであり,肺がんでは,システイン(C)に変異するG12C変異が最も高頻度に認められる.(この変異は面白いことにがん種によって異なり,大腸がん,すい臓がんではG12Dが最も多く,次いでG12Vとなり,G12Cは比較的低頻度である.すい臓がんではG12Rも多いが,他のがん種ではまれな変異タイプであり,このようながん種ごとの違いの背景にあるメカニズムは謎である.)KRASに対する阻害薬はさまざまなものが開発されてきたが十分な阻害活性が得られないままであった.しかし近年KRASのG12C変異体が「GDP結合状態」のときに一時的に薬剤が結合可能なポケットをSwitch II領域(アミノ酸番号では60~76残基あたり)に形成すること,マイケル付加反応を介して変異したシステインに共有結合可能な低分子化合物が開発されたことで,一気にKRASの低分子阻害薬研究が花開いた.現在までにソトラシブ,アダグラシブ(本邦は未承認)を含めて,複数のKRAS G12C阻害薬が開発されている.さらに,G12C以外の変異体に高い阻害活性を示す薬剤や,あらゆるKRASを阻害できる薬剤,KRASの活性化型に結合できる薬剤も開発され,臨床試験が進んでいる.
3)ALK阻害薬
現在までにわが国では5剤のALK阻害薬が承認され,さらに次世代のALK阻害薬の開発が続いている.
a.第一世代:クリゾチニブ
ALK融合遺伝子が発見されたときにMET阻害薬として臨床開発が始まっていた低分子阻害剤.METの阻害活性に加えて,ALKの阻害活性もあったことから,速やかにALK陽性肺がんへの臨床試験が開始され,最初のALK阻害薬となった.ALK, METだけでなくROS1への阻害活性も高く,現在ではROS1融合遺伝子を持つがんにも承認されて臨床応用されている.
b.第二世代:アレクチニブ,セリチニブ,ブリグチニブ
これらの第二世代阻害薬は,クリゾチニブに耐性を示す変異体にも有効性を示すことが多く,また,アレクチニブやブリグチニブは脳転移のある患者に対しても高い効果を示すとされる.中でもアレクチニブは,初回治療で使用した際のPFSの中央値が臨床試験において34か月を超え(クリゾチニブは9~11か月),さらに副作用が穏やかであるという特徴から,最も高頻度に初回治療薬として使われてきた.
c.第三世代:ロルラチニブ
脳への浸透力が高く,第二世代の薬に耐性が生じた場合にも有効とされ,あらゆる耐性変異にも高い有効性を示すとされる.脳転移巣への効果も非常に高い一方で,脳高次機能に対する可逆性の影響(副作用)が約4割強の患者で認められるというほかとは異なる特徴がある.初回治療からロルラチニブを使用した際のPFSは非常に長く,5年たってもまだ6割もの人が無増悪生存するという高い抗腫瘍効果が示されている.
4)ROS1阻害薬
ROS1はALKと非常に高いチロシンキナーゼ領域の相同性を持っており,いくつかのALK阻害薬がROS1阻害薬としても使用される.
a.クリゾチニブ
ALKに対するよりも低い濃度でROS1を阻害することができ,ROS1融合遺伝子陽性肺がんに使用された際のPFSはALK陽性肺がん患者に使用された際のPFSより長く(約19か月超),高い抗腫瘍効果が認められている.
b.エヌトレクチニブ
もともとNTRKの阻害薬としても開発されたが,ALKやROS1に対しても高い阻害活性を示し,ROS1融合遺伝子陽性肺がんの治療薬として承認されている.脳への移行性もよく,脳転移巣に対しても高い抗腫瘍効果が認められる.
c.レポトレクチニブなど
大環状構造を持つROS1阻害薬であり,クリゾチニブ耐性変異にも高い有効性を示す薬剤として開発され近年承認されている.他にもクリゾチニブ耐性変異にも有効な薬剤として,筆者らは以前タレトレクチニブ(DS6051b)を論文として報告しており24),現在第二相臨床試験までが行われ承認申請が行われている.
5)BRAF阻害薬
これまでに,最も頻度高く認められるBRAFのV600E変異に対しては,下記に記載の薬剤が承認されている(V600K変異も対象).実際の治療法としては,BRAF阻害のみではフィードバック経路が働き,CRAFを介したMEKの活性化が容易に起こるため,BRAF阻害薬とMEK阻害薬の併用療法が用いられる.また,肺がんなどで比較的よく見つかるV600E以外のBRAFクラスIIおよびクラスIII変異(非V600変異)に対する阻害薬の開発研究が加速しており,今後新たな阻害薬の登場が期待される.ダブラフェニブ,ベムラフェニブ,エンコラフェニブなどのBRAF阻害薬は,トラメチニブやコビメチニブといったMEK阻害薬との併用で使用されることがほとんどである.なお,BRAF変異陽性の大腸がんについては,BRAFを阻害するとEGFRの活性化を介したフィードバックが生じることから,抗EGFR抗体とエンコラフェニブの併用療法が承認されている.
6)RET阻害薬
RET融合遺伝子は肺がんの1~2%で発見されるが,RET特異的阻害薬が登場するまでは,RETを含むさまざまなチロシンキナーゼを阻害できるマルチキナーゼ阻害薬(バンデタニブ,カボザンチニブ,レンバチニブ)を用いた臨床試験が進められたが,承認には至らなかった.その後下記に示すRET選択的なRET-TKIが開発され,承認された.セルペルカチニブ,プラルセチニブ(本邦未承認)は,RETに対する選択的なチロシンキナーゼ阻害薬であり,RET融合遺伝子陽性肺がんに強力な増殖阻害活性を示し,持続的かつ高い治療効果が認められることから承認されて臨床応用されている.
7)その他
a.NTRK阻害薬:ラロトレクチニブ,エヌトレクチニブ
NTRK融合遺伝子陽性のがんに対して用いられ,肺がんに限らず,あらゆるがん種に有効とされ,がん種横断的使用が承認されている.
b.MET阻害薬:カプマチニブ,テポチニブ,グマロンチニブ
MET遺伝子のExon14スキッピング変異やMET遺伝子増幅は,がん細胞の増殖に関わる因子で,非小細胞肺がんの一部で発見される.その阻害薬として,これらの薬剤が承認されている.
上記で概説したように,喫煙者以外でも発症するタイプの肺がん(肺腺がん)においては,ドライバーがん遺伝子の同定とそれぞれのドライバーがん遺伝子産物への特異的阻害剤が開発され,進行がんであってもその予後の著しい改善がもたらされてきた.しかし,どれほど著効を示す分子標的薬でも,1年から数年の間にやがてがんが薬剤耐性を獲得し再増悪してしまうことが問題である.薬剤耐性細胞は,分子標的薬の治療が継続される中で,がん細胞がわずかに生き残り,それらが生き延びるための新たな遺伝子変異や耐性機構を獲得することで生じると考えられる.以下に,その薬剤耐性機構について,EGFR変異またはALK融合遺伝子陽性肺がんを中心に詳解する.
1)EGFR陽性肺がんの場合
EGFR変異肺がんにおける薬剤耐性機構は大きく次の三つに分けられる.
a.二次変異の獲得(T790M変異など)
第一世代EGFR阻害薬に対する耐性の約50~60%は,EGFR遺伝子のT790M変異によると報告されている.この変異は,EGFRの790番目のトレオニン(T)がメチオニン(M)に置換されるもので,ATPの結合部位に変化をもたらす25).1アミノ酸変異のみで耐性になる理由としては,T790M変異があると,EGFR受容体のATP結合部位が阻害薬よりもATPに親和性が高くなる一方で,阻害薬の親和性も低下する.そのため,EGFR阻害薬の効果が低下し,EGFR陽性がん細胞は治療薬存在下でも増殖できる.第二世代EGFR阻害薬に対してもT790M変異は耐性変異であることが知られている.
耐性克服療法としては,第三世代EGFR阻害薬(オシメルチニブ)が,T790M変異に対しても阻害活性を維持しており,T790M変異陽性の場合でも臨床上の有効性が認められている26).
第三世代EGFR阻害薬への耐性変異は,C797S変異が10%前後で認められる.797番目のシステイン残基は,オシメルチニブが共有結合する部位であるため,システイン残基でなくなることで,オシメルチニブ耐性となる.
b.バイパス経路の活性化(MET遺伝子増幅など)
EGFR自体には変異がなく,EGFR阻害薬によるEGFRからの増殖シグナルは阻害されているにもかかわらず,METなどのほかの遺伝子の活性化により,細胞生存シグナルが代償的に活性化されて耐性となる.比較的頻度高く認められるのが,MET遺伝子の増幅であり,また,リガンドであるHGF(hepatocyte growth factor)によりMET遺伝子の活性化が起こることによっても耐性になるとの報告もある27, 28).他にも,EGFRファミリーのHer2の遺伝子増幅などによる異常活性化もオシメルチニブ耐性機構として報告されている(オシメルチニブ耐性患者の5~10%に見つかる).METの遺伝子増幅はEGFR阻害薬による治療を受けている患者の約5~20%で見つかるとされており,MET阻害薬とEGFR阻害薬を併用することで耐性克服が可能とされているが(臨床研究段階),まだわが国ではバイパス経路による耐性を克服するための併用療法は承認されていない.
c.その他のメカニズムによる耐性
一部のEGFR変異陽性肺がんでは,治療を受けている過程で小細胞がんに形質転化し,EGFRからのシグナルに依存せずに生存・増殖するようになる症例が,約5~15%の患者に見つかる.このとき,EGFRの発現は低下し,ほとんどの症例でTP53の変異と,Rb遺伝子の機能喪失変異が見つかる29).この場合には,EGFR阻害薬にはまったく不応となり,いわゆる小細胞肺がんに対する化学療法(プラチナ製剤+エトポシド)が検討されることが多い.しかし,治療効果は限定的なことが多く,比較的予後不良となる.また,扁平上皮肺がんへと転化する症例もみられるが,確立した治療法はまだなく,転化の分子メカニズムも十分には明らかとなっていない.
EGFR以外の遺伝子(たとえばPIK3CA, MEK, BRAF, KRAS)などに新たな変異が生じて,EGFR阻害薬に対する耐性を獲得することしばしば生じる.実験的には新たに生じた活性化変異を抑制できる薬剤とEGFR阻害薬の併用が有用と考えられるが,確立した治療法はない.
また、がん細胞が上皮細胞から間葉系細胞に形態を変える「上皮間葉転換(EMT)」により,薬剤耐性が発生することがある.EMTが生じると,EGFR阻害薬への感受性が低下するとともに,より高い運動能を持ち,浸潤や転移能力が増すとされる.NFκB経路の活性化などが示唆されているが,不完全なEMTを起こす細胞でもEGFR阻害薬への耐性が認められており,詳細な耐性機構は不明な点も多い.この場合も確立した治療法は存在しない.
2)ALK融合遺伝子陽性肺がんの場合
ALK融合遺伝子陽性肺がんにおける薬剤耐性研究は,薬剤開発が進むと同時に進められてきた.第一世代,第二世代と薬剤が登場するにつれて,どのような耐性が生じ,どのような薬物療法によって耐性の克服が期待できるかといったことが研究されてきた.ALK融合遺伝子陽性肺がんでは,ある薬剤に耐性となった後でも,別のALK阻害薬が有効性を示すことが少なくないため,多くの患者ではこれまでに複数のALK阻害薬で逐次的に治療がなされてきた.その結果,平均生存期間は進行がんであっても5年以上であり,10年以上の生存が見込める症例も比較的多い.下記にEGFR同様耐性機構を分類して概説するが,各世代の阻害薬ごとに耐性機構が異なるため,阻害薬ごとに記載する.
a.クリゾチニブ(第一世代)への耐性
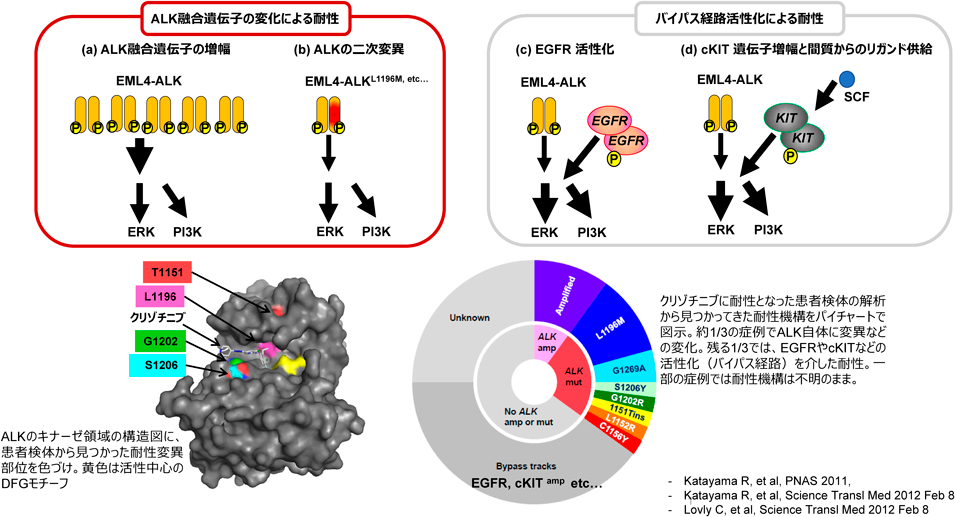
クリゾチニブはALK融合遺伝子を持つ非小細胞肺がんに対する治療薬として承認された初のALK阻害薬である.しかし,治療開始から1年以内で半数以上の症例で耐性が生じることが多い.その耐性機構としては,次の二つがあげられる.①ALK二次変異:クリゾチニブ耐性の約20~30%は,ALKタンパク質に新たな二次変異(L1196M, G1269A, C1156Y, G1202R, F1174Lなど)が見つかる.他には,ALK融合遺伝子の増幅により,治療抵抗性が生じることも報告されている.②バイパス経路の活性化:EGFRやc-KIT, IGF-1Rなどのほかのシグナル伝達経路が活性化し,ALK自身は阻害されていても,がん細胞が生存・増殖ができる状態になる(図1).なお,クリゾチニブはもともとMET阻害薬として開発されたということもあり,強力なMET阻害活性を有しているため,他のALK阻害薬で認められるようなMET遺伝子増幅を介した耐性が検出されないのが特徴である.
耐性克服法としては,クリゾチニブ耐性が確認された場合,第二世代のALK阻害薬(アレクチニブ,セリチニブ,ブリグチニブ)または第三世代ALK阻害薬(ロルラチニブ)へ切り替えることで治療効果が期待できることがある.
b.アレクチニブ,セリチニブ,ブリグチニブ(第二世代)への耐性
アレクチニブやセリチニブ,ブリグチニブは,クリゾチニブ耐性を示した患者に対しても一定の抗腫瘍効果が認められる.特徴としては,いずれもクリゾチニブよりも強力に(より低濃度で)ALKを阻害するだけでなく,G1202R以外のALK二次変異に対しても有効である.また,現在では,いずれの薬剤も初回治療で用いることも可能であり,実際にはアレクチニブが初回治療薬として最もよく使用されている.耐性機構としては,アレクチニブ耐性では,I1171T/N/S変異やV1180L変異,G1202R変異などが約3~4割の耐性患者において見つかる34).セリチニブに対してはF1174V/L変異やG1202R変異が35),ブリグチニブに対してはD1203N変異などが耐性変異とされたが,ブリグチニブはG1202R変異に対しては一定の効果が認められるとの報告がある(G1202Rを有する患者で奏効が得られたという報告がある)一方で,耐性患者からG1202R変異が見つかっている.また,次に述べるロルラチニブ同様,重複変異による耐性も課題となっている.バイパス経路の活性化を介した耐性については,クリゾチニブ耐性と同様,EGFRやFGFRなどのほかの受容体型チロシンキナーゼの活性化やその下流のPI3K/AKTやMAPKなどに関わる因子の活性化変異による耐性が知られている.クリゾチニブ耐性とは異なり,METの遺伝子増幅による耐性も一定頻度で認められる.その他の耐性機構として,セリチニブ耐性症例からは,ABCトランスポーターのP糖タンパク質(MDR1)が過剰発現する症例が見つかっており,P糖タンパク質の基質となるクリゾチニブやセリチニブには耐性となるものの,アレクチニブやロルラチニブには耐性とならない36).他には,低頻度であるが,EGFR変異肺がん同様に小細胞肺がんへの形質転化が認められる場合がある.
ALK遺伝子に耐性変異が見つかる場合には,耐性克服療法の候補としては,第三世代阻害薬のロルラチニブへの変更や,G1202R変異以外では,第二世代の阻害剤の中で薬剤を変更することでも持続的な抗腫瘍効果が認められることがある.たとえばアレクチニブ耐性変異のI1171N/S/TやV1180L変異はブリグチニブに高感受性を示す.
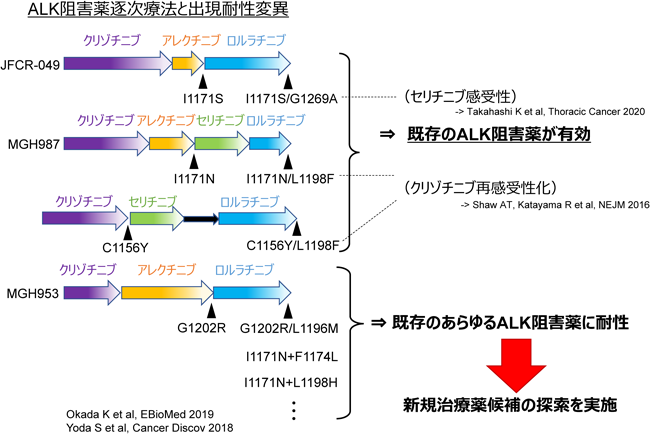
c.ロルラチニブ(第三世代)への耐性
第三世代ALK阻害薬のロルラチニブは第一世代のクリゾチニブと基本骨格は同じままで大環状構造(12員環)にした阻害薬であり,これまで患者で見つかってきたあらゆるALKの耐性変異(重複変異は除く)にも高い阻害活性を示すため,第二世代阻害薬に耐性を示した患者で,ALKの耐性変異がある場合には高い抗腫瘍効果が期待できる37).また,脳脊髄液中への移行が非常によく,脳転移に対する効果も非常に高い.その耐性機構としては,ロルラチニブが二次治療以降に使用されることが多かったため,二つ以上の変異が一つのALKタンパク質に生じる重複変異があげられる.たとえば,C1156Y+L1198F変異が最初にロルラチニブ耐性の重複変異として報告されたが,二つ目の変異が入ることで,著しくロルラチニブの阻害活性が低下する.このような重複変異の組合わせは非常に種類が多く,二次治療以降としてロルラチニブが使用された患者の4割以上で重複耐性変異が報告されている38, 39).非常に興味深い点としては,これら重複変異の一部は,もともと耐性であった第一,第二世代ALK阻害薬に再感受性を示す場合があることである.たとえば,ALK-C1156Y変異体はクリゾチニブに高度耐性を示すものの,C1156Y+L1198Fはロルラチニブ耐性を示すが,クリゾチニブに再度感受性化するようになる40),などである(図2).最近,ロルラチニブを初回治療から使用した際のPFSが非常に長いことが報告されてから,ロルラチニブを初回治療として使用するケースも少なくない.そのため,実際にはロルラチニブ初回治療後に出てくる耐性はどのようなものがあるかといったことが今後解明される必要がある.その他のALK阻害薬耐性機構としては,第一,第二世代ALK阻害薬同様,バイパス経路の再活性化がMETの遺伝子増幅やPI3K, MAPKなどの活性化,NF2の欠損などで生じることが報告されている.
なお,ロルラチニブ耐性が,既存薬への再感受性化が期待される変異であった場合には,第一,第二世代のALK阻害薬による抗腫瘍効果も期待できるが,ALK阻害薬でなく,従来からのがん化学療法による治療なども行われる.我々の研究室をはじめ,世界中でさまざまな次世代ALK阻害薬が探索・研究開発されており,いくつかのあらゆる既存ALK阻害薬に対して耐性を示す重複変異体にも有効性を示す化合物についての臨床試験が進んでいる.今後それらの有効性が示されて治療の選択肢がさらに広がることを期待したい(図3).
3)ALK阻害薬耐性機構のまとめ
ALK融合遺伝子陽性肺がんにおける薬剤耐性は,使用する薬剤の種類(第一世代,第二世代,第三世代)によって異なるメカニズムが関与する.特に,クリゾチニブから第二世代,第三世代への移行に伴い,ALKに生じる二次変異やバイパス経路の活性化,複合変異などの複雑なメカニズムが認められる.各世代の薬剤で耐性が確認された場合には,次世代の薬剤や異なる経路を標的とする薬剤の併用などが考慮され,個別の耐性機構に合わせた治療の開発が期待されるが,症例数が限られることもあり有効性を検証するための臨床研究は非常に困難である.
肺がんの分子標的薬に対する耐性研究は,分子標的薬が登場したときから課題となって顕在化し,これまで世界中で数多くの研究者がこの課題に取り組んできた.2005年にEGFR-T790Mが報告されてからまもなく20年となるが,いまだ薬剤耐性との戦いは終わりがみえない.とはいえ,この約20年の間に,数多くの耐性機構の発見とその克服法候補が発見されてきた.ここではいまだ解明できていないメカニズムと,さらなる治療抵抗性克服のための研究戦略について概説したい.
1)いまだに不明な耐性機構
EGFR変異陽性肺がんやALK融合遺伝子陽性肺がんでは,耐性時の検体や血液検体を用いた薬剤耐性機構の探索が行われ,薬剤の標的遺伝子におけるさまざまな二次変異やバイパス経路活性化機構などが判明してきた.薬剤やドライバーがん遺伝子の違いによって,差異はあるものの,基本的な耐性機構のパターンや種類は高い類似性がある.しかし,約2~3割のケースでは,耐性機構がいまだ不明なままである.筆者の所属する研究施設においても,これまでに,病院と研究所の連携により,倫理審査委員会承認済みのプロトコールに則り,同意をいただいた患者の検体を用いた研究を行ってきた.特に,薬物治療に抵抗性となった際に,病勢進行に伴って貯留した胸水や心嚢水,腹水を排液した際の体腔液や,臨床検査のための生検の残余検体を用いて治療抵抗性機構の探索を行ってきた.これまでに,500症例以上から,さまざまな治療抵抗性機構を探索・同定してきたが,どうしてもメカニズムが不明な症例が残っている.我々は,可能な限りいただいた新鮮臨床検体から培養細胞株を樹立し,新鮮検体由来のDNAやRNAを用いた遺伝子変異・発現解析だけでは明らかにできないメカニズムの探索に挑戦している.標的遺伝子に遺伝子変異がある場合や明らかなバイパス経路活性化の遺伝子異常がある場合は(新規変異であれば検証実験を行うが)耐性機構判明ということで,その検体を用いた解析はいったん終了となる(もちろん培養細胞株化して,将来的な治療抵抗性に関わる基礎研究には活用させていただく).しかし,明らかに抵抗性に関わる変異が見いだせない場合は,培養細胞株にしてさらなる検証を行う.もともとの分子標的薬と,標的既知の薬剤を併用するスクリーニングから抵抗性細胞で活性化しているシグナルを推定する方法や,最近では,約3000種の化合物を用いたスクリーニングやCRISPRスクリーニングといった方法も実施し,抵抗性機構の解明に努めている.しかし,そのような検討には非常に時間がかかる上,同定したメカニズムが実際どのような変化(発現変化や遺伝子変異など)が起点となって耐性につながったのか,といったところまで明らかにするには,さらに多くの時間を要する.また,腫瘍微小環境で初めて耐性が維持されるといった例も少なくない上,腫瘍細胞以外の細胞から出される因子が耐性に関わるといった可能性も検討する必要がある.そのためには,培養条件も通常の2次元培養だけでは,耐性機構の同定につながらないかもしれない.耐性時に,検査をせずに次の治療選択に進む場合も多いと思われるため,なかなか耐性時の検体を新鮮な条件で培養するというのは難しいかもしれないが,可能な限り培養できる新鮮な状態で研究に利用することで,徐々に未同定の耐性機構が解明できるようになるかもしれない.
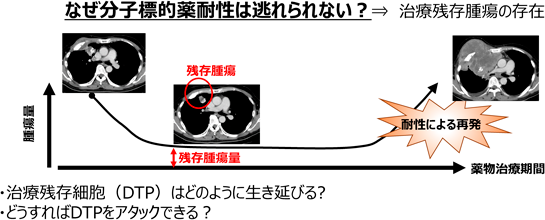
2)治療残存細胞(DTP細胞)
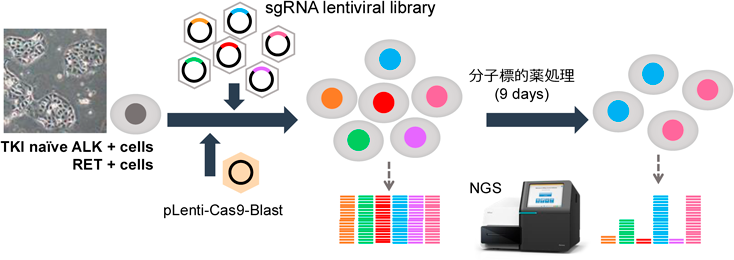
がん分子標的薬耐性の出現が,ほとんどの症例で起こってしまうのは,そもそも治療がどれほど劇的に効いてもごくわずかに生き延びるがん細胞がいるからにほかならない.以前は,このような治療抵抗性をもって残存する細胞を「がん幹細胞」として解析する研究が2000年ごろから盛んに行われるようになっていたが,近年ではこれらの治療で生き延びる細胞を「治療残存(drug tolerant persister:DTP)細胞」と呼ぶことが多い.このDTP細胞がどうして生き延びるのか,どのような性質を持ち,どのようにしてこの生き延びた細胞から耐性細胞が出現してくるのか,といったことが盛んに研究されるようになった(図4).2011年に培養実験で肺がんのDTP細胞研究の礎とも呼ぶ論文が発表され,今でもその研究手法が踏襲されてDTP研究が行われている.なお,そのDTP細胞に関する研究は,筆者が留学中にLabセミナーを一緒に行っていた隣の研究室(PI:Jeff Settleman博士)からCell誌に発表された論文であり,留学開始間もないころだった自分は,周りの研究者らのレベルに圧倒されたのを覚えている41).さて,このDTP細胞であるが,近年ではさまざまな解析手法を用いてその性状解析が行われている.たとえばDTP細胞とその親株の発現遺伝子をRNAシーケンスなどで比較する,1細胞解析でどのような細胞がDTPになっていくのか,エピゲノムの変化はどうなっているか,なぜDTPになる細胞は薬剤存在下で死なずに生き延びられるのか(細胞周期は?)などの視点で,世界中で研究が進められている.筆者らは,CRISPR-Cas9システムを用いたゲノムワイドのノックアウトスクリーニングを行い,どのような遺伝子の発現が喪失するとDTPとして残りやすいか,という検討をクローン化したCas9発現細胞(原則的に同一の遺伝子変異のバックグラウンドを持っていると考えられる細胞)を用いて行っている(図5).たとえば,ERRFI1(MIG6)の欠損は,きわめて微量のEGFRリガンド(血中濃度以下のEGFなど)でEGFRの活性化を起こせるようになり,容易に耐性を誘導する42, 43)(図6).これ以外にも複数のDTP生存メカニズムを見いだすことに成功し,現在もさまざまな因子について解析を進めている.DTPの研究からみえてくることは,DTPとして残る細胞もきわめて多様なメカニズムで生き延びていることである.このことからも,生物の持つ「過酷な状況でも生き延びてきた可塑性」を,がん細胞は巧みに利用して生き延びているということを痛感する.
3)治療残存細胞から耐性細胞への進化
先にDTP細胞がさまざまなメカニズムで生き延びていることを概説したが,この生き延びた細胞もそのままでは薬剤存在下でなかなか爆発的に増殖できるわけではなく,薬剤による増殖シグナル阻害の影響を受けて半ば休眠状態のような形で生きている.では,このDTP細胞が,どのようにして完全な耐性細胞(薬剤があってもまったく気にすることなく増殖できる耐性細胞)に“進化”するのか,が解明すべき点である.もともとがんの発生時に,「がんの進化(悪性化)」についての研究は盛んに行われてきたが,薬剤耐性細胞への“進化”という視点での研究が最近盛んに進められるようになってきている.
DTP細胞から耐性細胞への進化を耐性機構から考えると,何らかの遺伝子変異や遺伝子構造異常(遺伝子増幅)などにより,バイパス経路が活性化される場合と,標的遺伝子に耐性変異(EGFRのT790M変異やALKのG1202R変異など)が獲得される場合に大きく分けることができる.EGFR変異肺がんでは,先に述べたようにEGFR-T790Mが第一,第二世代EGFR阻害薬への耐性機構の5割以上を占めるが,なぜこの変異ばかりが高頻度に生じるのかについては永らく謎であった.最近になりDNAのデアミナーゼの過剰発現が特定の変異を加速しうることが報告されている.たとえば,EGFR変異肺がんにおいて,分子標的薬(EGFR阻害薬)で処理することで,NFκBなどを介して,一本鎖核酸のCをU/Tに変換するデアミナーゼであるAPOBEC3A(A3A)が誘導されること,EGFR阻害薬耐性腫瘍では,APOBECを介して生じるとされる遺伝子変異パターン(変異シグニチャー)が増えていることが報告された44).ほぼ同時期に別のグループからは,同様にNFκBを介したAPOBEC3B(A3B)の発現上昇が,変異誘導や遺伝子構造異常の誘導に関わり,EGFR阻害薬耐性に寄与している可能性が示唆された.一方,発がん段階においては,A3Bの発現はEGFR変異肺がんの腫瘍形成を抑制することも示され,非小細胞肺がんにおけるA3Bの多面的な役割が明らかにされている44, 45).これらの研究からA3AやA3BといったAPOBECを標的とすることで,より持続的な分子標的薬の効果をもたらす可能性が示されたと考えられるが,まだ直接的になぜ特定の遺伝子変異が生じやすいかといったことまでは明らかにできているとはいえず,今後さらなる研究が必要である.
4)克服すべき課題
現時点では結局,耐性細胞の克服薬を見つけてもさらなる耐性が生じ,という「いたちごっこ」から抜け出せていない.ここを抜け出すためのストラテジーを発見する必要がある.また,多剤耐性になった細胞は高い可塑性(柔軟な対応能力)を備えており,治療が困難となっている.しかしこのような細胞にもまったく別の視点での研究から新たな脆弱性を見いだすことができるかもしれないが,まだまだ研究が必要である.また,腫瘍微小環境を模倣し,免疫細胞など宿主のほかの細胞を考慮した実験系の再構築も,今後より重要な課題となってくると考えられる.
がん免疫チェックポイント阻害薬(immune checkpoint inhibitor:ICI),特にPD-1(programmed cell death-1)/PD-L1およびCTLA-4(cytotoxic T-lymphocyte antigen-4)阻害薬などは,免疫系を再活性化してがん細胞を攻撃させる治療法である.しかし,がん細胞はそもそも免疫からの監視をかいくぐって増大してきただけでなく,ICI治療に適応し,さまざまなメカニズムでICI抵抗性を獲得する.ICIへの耐性も,初期耐性(治療開始から効果が現れない)と獲得耐性(治療効果がいったんみられたのちに再発する)に分けられる.ICI治療のみでは,奏効する割合が約2~3割とされており,初期耐性が非常に多い.さらに,一定の効果がみられた場合でも,獲得耐性が生じて耐性となる症例も少なくない.以下に,代表的なICI耐性機構を簡単に概説する46).
1)免疫原性の低下
がん細胞が免疫原性を欠く場合,T細胞が標的として認識しにくくなり,ICIの治療効果が期待できない.たとえば,がん細胞がMHCクラスI分子の発現を低下させたり,MHCクラスI分子を細胞表面に提示するために必要なβ2ミクログロブリン遺伝子の欠損が生じたりすると,抗原性(抗原提示)が低下し,T細胞ががん細胞を認識できなくなる.また,がん抗原そのものが少なくなる,または発現喪失することでも,がん細胞は免疫監視から逃れてしまう.
2)チェックポイント分子(PD-L1)の再発現
免疫チェックポイント阻害薬がPD-1/PD-L1経路をブロックしても,がん細胞がPD-L1の発現量を再び上昇させて薬剤効果を回避することがある.そのメカニズムとしては,がん細胞や腫瘍周囲の免疫抑制性細胞がINF-γシグナルを介してPD-L1の発現をさらに増加させ,T細胞の機能を抑えることがある.また,分泌型PD-L1を発現するようになることで,抗PD-L1抗体をトラップしてしまい,耐性を引き起こす場合があることも報告されている47).
3)抑制性免疫細胞の浸潤が増加
腫瘍組織内のがん細胞の周囲に制御性T細胞(Treg)などの免疫抑制性細胞が集積することで,T細胞の攻撃性が抑制される.Treg以外にも,骨髄由来抑制性細胞,腫瘍関連マクロファージなどががん微小環境に集積し,T細胞の活性化を抑制するサイトカインなどを分泌することでT細胞の機能を低下させる.
4)代謝環境の変化
腫瘍微小環境が低酸素・低pH・低グルコースなどの過酷な状態になると,解糖系を主に使用して活性化・増殖する細胞障害性T細胞はエネルギーを消耗し,機能が低下する.一方で,本来炎症などの免疫応答が起きた後の「火消し役」として働くTregなどは,過酷な条件下(高乳酸など)で生き延びられる細胞内代謝系を有しており,免疫抑制的な腫瘍微小環境が作られる.
5)新しい免疫チェックポイント分子の発現
がん細胞や周囲の免疫抑制性細胞がPD-1/PD-L1とは異なる新しいチェックポイント分子を発現し,T細胞の機能を抑える.たとえば,T細胞の機能抑制に関わる分子であるTIGIT(T-cell immunoreceptor with Ig and ITIM domains),LAG-3(lymphocyte-activation gene 3),TIM-3(T cell immunoglobulin and mucin domain-containing protein 3)が発現することで,T細胞の機能が抑制される.
6)シグナル伝達経路の変化による免疫回避経路の変化
がん細胞がJAK1/2の変異を獲得することで,INF-γ受容体からのシグナル伝達経路が阻害され,PD-L1発現が減少するなどの免疫不応答な腫瘍環境が形成され,ICI耐性が誘導される.
このように,がん免疫チェックポイント阻害薬に対する耐性機構は多様であり,がん細胞自身の変化だけでなく,腫瘍微小環境の変化によっても複合的に抵抗性が獲得される.これらのICI耐性を克服するためには,絶妙なバランスの上で成り立っている免疫応答に対してがん細胞が抵抗性を獲得するメカニズムを詳細に理解することが必要である.それらの耐性獲得のメカニズムに対抗する複数の治療方法を各患者に合わせて適用することで,腫瘍免疫応答を強化することが可能になると考えられる.現在開発が進むワクチンや改変型T細胞など,さまざまな新しい技術が加わることで,より効果的ながん免疫療法の確立が期待できる.
~分子標的薬耐性研究との忘れられない出会い~
筆者は,2006年3月に博士課程を卒業後,がん研究会にて「がん幹細胞についての研究」や「がんの増殖および細胞死に関わる因子の基礎研究」を行っていたが,2010年度に日本学術振興会の海外特別研究員にご採択いただき,2010年3月に渡米し,マサチューセッツ総合病院(MGH)がんセンターにて留学をさせていただいた.当時は,2007年に間野博士らのグループによりNature誌に発表されたEML4–ALK融合遺伝子陽性肺がんに対するクリゾチニブの臨床試験が,米国では留学先のMGHを中心として行われていた.留学した研究室のボスであったJeffrey Engelman博士は呼吸器内科医であり,MGHがんセンターの呼吸器内科にてAlice Shaw博士をPIとして行われていたクリゾチニブの臨床試験30)で耐性となった患者の解析がEngelman Labで行われようとしていた.そこに飛んで火に入ったのが筆者であり,留学を開始するや否や,ALKの耐性研究プロジェクトの開始が命じられた.すぐさま,実験的な耐性細胞の樹立とともに,患者検体を受け取るたびに培養株の樹立が命じられ,目の回るような忙しさとなった.ボスとのMeetingは毎週行われ,そのMeetingにはほどなくしてShaw博士も参加するようになり,当時Shaw博士が雇用していたテクニシャン(米国では大学を卒業してから,著名なLabでテクニシャンをしてから“いい推薦書”を手にメディカルスクールに挑戦するというのが多く,Labにはそのような若いテクニシャンが複数在籍していた)も一緒になってALK阻害薬耐性研究プロジェクトが推進された.また,それまではまだ作られていなかったPDX(patient-derived xenografts;患者腫瘍組織移植)モデルの作製もすることになった(動物実験を担当しているLabマネージャーがおられたが,時間外での勤務対応は当然ながら米国では無理であり,臨床検体が出る時間が少しでも遅い場合には自らで行うことが多かった.さらに運悪く(?)ちょうどワールドカップサッカー2010(南アフリカにて6月中旬から7月に開催)の年でもあり,ナイターで行われるワールドカップの試合はちょうどアメリカでは昼(6時間の時差)の放送であったため,マウスの実験は自らで行うか,同じLabにいた日本人の先輩研究者(現在愛知県がんセンターでご活躍の衣斐先生)にご協力いただくことが必須であった.おかげさまで,PDXの樹立や継代・維持,そして胸水や生検などの臨床検体からの初代培養技術確立も自分のスキルとなり,2012年夏に帰国してからの研究で,自分の強力な武器となった.耐性細胞作りと患者検体を用いた耐性機構探索研究は,毎週のボスからの強力なプッシュと,Shaw博士からの,「あの患者さんの耐性メカニズム何かわかった?」という悪気のない強烈なプレッシャーにより,ありがたいくらい順調に進んだ.留学開始から半年がたったころ,研究に転機が訪れた.留学についてきてくれた家内とともにアメリカ国内を1週間弱旅行する夏休みをとったのだが,戻ってくると明らかに樹立中の耐性細胞の「顔つき」が変わっていた.まったく薬剤の存在を気にしない「顔」になっていた.すぐさま,ALK融合遺伝子をシーケンス解析したところ,変異があることがわかった.それがL1196Mゲートキーパー変異[ATP結合ポケットの奥にあたるヒンジ部を構成するアミノ酸(TやLなど)が,Mなどに変化することで薬剤親和性が低下し,代わりにATPとの親和性が増強することで,耐性変異となる]であった.EGFRのT790Mに相当する位置のアミノ酸が見事に変異を起こしていた.発見当時はまだ報告がなかったため,ボスと相当興奮したことを覚えている.しかしその興奮はすぐさま打ち砕かれることとなった.日本から間野博士らのグループが2010年10月28日号のNEJM誌に,世界初のクリゾチニブ耐性変異として同じ変異を発見され,報告されたのである31).留学して半年,コツコツ築いてきた成果が散った瞬間であった.しかし,ボスはそこでは終わらず,L1196M耐性を克服できる薬剤を開発するため,当時ALK阻害薬の開発を行っていたAriad社(2017年に武田薬品により買収)との共同研究を開始し,迅速に論文化までもっていく作戦となった.当時はようやく英会話に少しはついていけるようにはなっていたが,まだ右も左もわからないまま,Ariad社の社員の方に自らの研究をプレゼンさせられ,あれよあれよという間に共同研究が開始し,化合物(現在のブリグチニブ)があっという間に届いた.予想どおりに,L1196Mゲートキーパー変異への阻害活性があることを,培養株,動物実験いずれでも示すことができ,2010年12月には投稿し,リバイズを経て,翌年3月末にはPNASにアクセプトとなり,留学をしに来た身としてはまずは一安心したのを覚えている32).しかしその間もクリゾチニブ耐性の臨床検体は次から次へと出てくるため,次第に検体からの培養を手伝ってくれるメンバー(テクニシャン)が増えた.なお,一緒に働いた二人のテクニシャンは無事メディカルスクールに合格し,今では米国で外科医,内科医として活躍されている.
留学2年目には,クリゾチニブ耐性症例を約20症例詳細に解析し,新たな抵抗性メカニズムを複数同定して論文とすることができた.この論文は,自らの力だけでは成し遂げることが決してできなかった研究であると痛感している.臨床医,病理医,分子病理医(今でいうNGS解析をも行う病理医),研究者,テクニシャンがチームとなって協力してくださったおかげで,非常に迅速に1本の論文(自分にとっては大作)に仕上げることができた.この論文はALK耐性機構の礎となる論文ととらえていただいており,これまでに1400回を超えて引用していただいている33).
米国留学では,日本以上にチームで互いが得意とする技術を最大限出し合い,ものすごくスピーディーに研究が推進していくさまを目のあたりにすることができた.このようなチームでの研究を日本でもしたいと思い,2012年夏にがん研究会に帰国後さまざまな先生にご協力をいただきながら今までやってこられたと思う一方,この4年間のコロナ禍は,協力体制の維持継続,特に新規構築が,非常に難しいと感じる時期でもあった.
引用文献References
1) Aberle, D.R., Adams, A.M., Berg, C.D., Black, W.C., Clapp, J.D., Fagerstrom, R.M., Gareen, I.F., Gatsonis, C., Marcus, P.M., & Sicks, J.D.; National Lung Screening Trial Research Team. (2011) Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med., 365, 395–409.
2) Sordella, R., Bell, D.W., Haber, D.A., & Settleman, J. (2004) Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science, 305, 1163–1167.
3) Saito, M., Shiraishi, K., Kunitoh, H., Takenoshita, S., Yokota, J., & Kohno, T. (2016) Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci., 107, 713–720.
4) Rodenhuis, S., van de Wetering, M.L., Mooi, W.J., Evers, S.G., van Zandwijk, N., & Bos, J.L. (1987) Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N. Engl. J. Med., 317, 929–935.
5) Soda, M., Choi, Y.L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., Fujiwara, S., Watanabe, H., Kurashina, K., Hatanaka, H., et al. (2007) Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature, 448, 561–566.
6) Mano, H. (2012) ALKoma: A cancer subtype with a shared target. Cancer Discov., 2, 495–502.
7) Kiyozumi, D., Noda, T., Yamaguchi, R., Tobita, T., Matsumura, T., Shimada, K., Kodani, M., Kohda, T., Fujihara, Y., Ozawa, M., et al. (2020) NELL2-mediated lumicrine signaling through OVCH2 is required for male fertility. Science, 368, 1132–1135.
8) Rikova, K., Guo, A., Zeng, Q., Possemato, A., Yu, J., Haack, H., Nardone, J., Lee, K., Reeves, C., Li, Y., et al. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell, 131, 1190–1203.
9) Bergethon, K., Shaw, A.T., Ou, S.H., Katayama, R., Lovly, C.M., McDonald, N.T., Massion, P.P., Siwak-Tapp, C., Gonzalez, A., Fang, R., et al. (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol., 30, 863–870.
10) Davies, K.D., Le, A.T., Theodoro, M.F., Skokan, M.C., Aisner, D.L., Berge, E.M., Terracciano, L.M., Cappuzzo, F., Incarbone, M., Roncalli, M., et al. (2012) Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res., 18, 4570–4579.
11) Takeuchi, K., Soda, M., Togashi, Y., Suzuki, R., Sakata, S., Hatano, S., Asaka, R., Hamanaka, W., Ninomiya, H., Uehara, H., et al. (2012) RET, ROS1 and ALK fusions in lung cancer. Nat. Med., 18, 378–381.
12) Davies, H., Bignell, G.R., Cox, C., Stephens, P., Edkins, S., Clegg, S., Teague, J., Woffendin, H., Garnett, M.J., Bottomley, W., et al. (2002) Mutations of the BRAF gene in human cancer. Nature, 417, 949–954.
13) Brose, M.S., Volpe, P., Feldman, M., Kumar, M., Rishi, I., Gerrero, R., Einhorn, E., Herlyn, M., Minna, J., Nicholson, A., et al. (2002) BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res., 62, 6997–7000.
14) Nieto, P., Ambrogio, C., Esteban-Burgos, L., Gomez-Lopez, G., Blasco, M.T., Yao, Z., Marais, R., Rosen, N., Chiarle, R., Pisano, D.G., et al. (2017) A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature, 548, 239–243.
15) Yao, Z., Yaeger, R., Rodrik-Outmezguine, V.S., Tao, A., Torres, N.M., Chang, M.T., Drosten, M., Zhao, H., Cecchi, F., Hembrough, T., et al. (2017) Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature, 548, 234–238.
16) Kohno, T., Ichikawa, H., Totoki, Y., Yasuda, K., Hiramoto, M., Nammo, T., Sakamoto, H., Tsuta, K., Furuta, K., Shimada, Y., et al. (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat. Med., 18, 375–377.
17) Drilon, A., Oxnard, G.R., Tan, D.S.W., Loong, H.H.F., Johnson, M., Gainor, J., McCoach, C.E., Gautschi, O., Besse, B., Cho, B.C., et al. (2020) Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N. Engl. J. Med., 383, 813–824.
18) Izumi, H., Matsumoto, S., Liu, J., Tanaka, K., Mori, S., Hayashi, K., Kumagai, S., Shibata, Y., Hayashida, T., Watanabe, K., et al. (2021) The CLIP1-LTK fusion is an oncogenic driver in non-small-cell lung cancer. Nature, 600, 319–323.
19) Cancer Genome Atlas Research Network. (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature, 489, 519–525.
20) Janne, P.A., Yang, J.C., Kim, D.W., Planchard, D., Ohe, Y., Ramalingam, S.S., Ahn, M.J., Kim, S.W., Su, W.C., Horn, L., et al. (2015) AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med., 372, 1689–1699.
21) Soria, J.C., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K.H., Dechaphunkul, A., Imamura, F., Nogami, N., Kurata, T., et al.; FLAURA Investigators. (2018) Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med., 378, 113–125.
22) Yun, J., Lee, S.H., Kim, S.Y., Jeong, S.Y., Kim, J.H., Pyo, K.H., Park, C.W., Heo, S.G., Yun, M.R., Lim, S., et al. (2020) Antitumor activity of amivantamab (JNJ-61186372), an EGFR–MET bispecific antibody, in diverse models of EGFR exon 20 insertion-driven NSCLC. Cancer Discov., 10, 1194–1209.
23) Park, K., Haura, E.B., Leighl, N.B., Mitchell, P., Shu, C.A., Girard, N., Viteri, S., Han, J.Y., Kim, S.W., Lee, C.K., et al. (2021) Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: Initial results from the CHRYSALIS Phase I study. J. Clin. Oncol., 39, 3391–3402.
24) Katayama, R., Gong, B., Togashi, N., Miyamoto, M., Kiga, M., Iwasaki, S., Kamai, Y., Tominaga, Y., Takeda, Y., Kagoshima, Y., et al. (2019) The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat. Commun., 10, 3604.
25) Kobayashi, S., Boggon, T.J., Dayaram, T., Janne, P.A., Kocher, O., Meyerson, M., Johnson, B.E., Eck, M.J., Tenen, D.G., & Halmos, B. (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med., 352, 786–792.
26) Cross, D.A., Ashton, S.E., Ghiorghiu, S., Eberlein, C., Nebhan, C.A., Spitzler, P.J., Orme, J.P., Finlay, M.R., Ward, R.A., Mellor, M.J., et al. (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov., 4, 1046–1061.
27) Engelman, J.A., Zejnullahu, K., Mitsudomi, T., Song, Y., Hyland, C., Park, J.O., Lindeman, N., Gale, C.M., Zhao, X., Christensen, J., et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science, 316, 1039–1043.
28) Yano, S., Wang, W., Li, Q., Matsumoto, K., Sakurama, H., Nakamura, T., Ogino, H., Kakiuchi, S., Hanibuchi, M., Nishioka, Y., et al. (2008) Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res., 68, 9479–9487.
29) Niederst, M.J., Sequist, L.V., Poirier, J.T., Mermel, C.H., Lockerman, E.L., Garcia, A.R., Katayama, R., Costa, C., Ross, K.N., Moran, T., et al. (2015) RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun., 6, 6377.
30) Shaw, A.T., Yeap, B.Y., Solomon, B.J., Riely, G.J., Gainor, J., Engelman, J.A., Shapiro, G.I., Costa, D.B., Ou, S.H., Butaney, M., et al. (2011) Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol., 12, 1004–1012.
31) Choi, Y.L., Soda, M., Yamashita, Y., Ueno, T., Takashima, J., Nakajima, T., Yatabe, Y., Takeuchi, K., Hamada, T., Haruta, H., et al.; ALK Lung Cancer Study Group. (2010) EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med., 363, 1734–1739.
32) Katayama, R., Khan, T.M., Benes, C., Lifshits, E., Ebi, H., Rivera, V.M., Shakespeare, W.C., Iafrate, A.J., Engelman, J.A., & Shaw, A.T. (2011) Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA, 108, 7535–7540.
33) Katayama, R., Shaw, A.T., Khan, T.M., Mino-Kenudson, M., Solomon, B.J., Halmos, B., Jessop, N.A., Wain, J.C., Yeo, A.T., Benes, C., et al. (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med., 4, 120ra17.
34) Katayama, R., Friboulet, L., Koike, S., Lockerman, E.L., Khan, T.M., Gainor, J.F., Iafrate, A.J., Takeuchi, K., Taiji, M., Okuno, Y., et al. (2014) Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res., 20, 5686–5696.
35) Friboulet, L., Li, N., Katayama, R., Lee, C.C., Gainor, J.F., Crystal, A.S., Michellys, P.Y., Awad, M.M., Yanagitani, N., Kim, S., et al. (2014) The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov., 4, 662–673.
36) Katayama, R., Sakashita, T., Yanagitani, N., Ninomiya, H., Horiike, A., Friboulet, L., Gainor, J.F., Motoi, N., Dobashi, A., Sakata, S., et al. (2016) P-glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase-rearranged non-small cell lung cancer. EBioMedicine, 3, 54–66.
37) Zou, H.Y., Friboulet, L., Kodack, D.P., Engstrom, L.D., Li, Q., West, M., Tang, R.W., Wang, H., Tsaparikos, K., Wang, J., et al. (2015) PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell, 28, 70–81.
38) Yoda, S., Lin, J.J., Lawrence, M.S., Burke, B.J., Friboulet, L., Langenbucher, A., Dardaei, L., Prutisto-Chang, K., Dagogo-Jack, I., Timofeevski, S., et al. (2018) Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov., 8, 714–729.
39) Okada, K., Araki, M., Sakashita, T., Ma, B., Kanada, R., Yanagitani, N., Horiike, A., Koike, S., Oh-Hara, T., Watanabe, K., et al. (2019) Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine, 41, 105–119.
40) Shaw, A.T., Friboulet, L., Leshchiner, I., Gainor, J.F., Bergqvist, S., Brooun, A., Burke, B.J., Deng, Y.L., Liu, W., Dardaei, L., et al. (2016) Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N. Engl. J. Med., 374, 54–61.
41) Sharma, S.V., Lee, D.Y., Li, B., Quinlan, M.P., Takahashi, F., Maheswaran, S., McDermott, U., Azizian, N., Zou, L., Fischbach, M.A., et al. (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell, 141, 69–80.
42) Kondo, N., Utsumi, T., Shimizu, Y., Takemoto, A., Oh-Hara, T., Uchibori, K., Subat-Motoshi, S., Ninomiya, H., Takeuchi, K., Nishio, M., et al. (2023) MIG6 loss confers resistance to ALK/ROS1 inhibitors in NSCLC through EGFR activation by low-dose EGF. JCI Insight, 8, e173688.
43) Wei, X., Uchibori, K., Kondo, N., Utsumi, T., Takemoto, A., Koike, S., Takagi, S., Yanagitani, N., Nishio, M., & Katayama, R. (2024) MIG6 loss increased RET inhibitor tolerant persister cells in RET-rearranged non-small cell lung cancer. Cancer Lett., 604, 217220.
44) Isozaki, H., Sakhtemani, R., Abbasi, A., Nikpour, N., Stanzione, M., Oh, S., Langenbucher, A., Monroe, S., Su, W., Cabanos, H.F., et al. (2023) Therapy-induced APOBEC3A drives evolution of persistent cancer cells. Nature, 620, 393–401.
45) Caswell, D.R., Gui, P., Mayekar, M.K., Law, E.K., Pich, O., Bailey, C., Boumelha, J., Kerr, D.L., Blakely, C.M., Manabe, T., et al. (2024) The role of APOBEC3B in lung tumor evolution and targeted cancer therapy resistance. Nat. Genet., 56, 60–73.
46) Schoenfeld, A.J. & Hellmann, M.D. (2020) Acquired resistance to immune checkpoint inhibitors. Cancer Cell, 37, 443–455.
47) Gong, B., Kiyotani, K., Sakata, S., Nagano, S., Kumehara, S., Baba, S., Besse, B., Yanagitani, N., Friboulet, L., Nishio, M., et al. (2019) Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J. Exp. Med., 216, 982–1000.
著者紹介Author Profile
 片山 量平(かたやま りょうへい)
片山 量平(かたやま りょうへい)公益財団法人がん研究会がん化学療法センター基礎研究部 部長.博士(薬学).
略歴2001年東大薬学部卒業,06年東大・院・薬学博士課程修了・学位(薬学博士)取得.同年より癌研究会癌化学療法センター基礎研究部研究員.学振海外特別研究員として米国留学(10~12年)を経て,現所属に研究員として復職.主任研究員を経て17年より現職.
研究テーマと抱負肺がんの分子標的治療薬耐性機構に関する研究,がん免疫チェックポイント阻害薬の感受性・抵抗性規定因子の研究を中心に,大腸がんや骨肉腫,グリオブラストーマなどに対する新規治療標的の探索研究も展開している.いつか自分たちの発見が臨床に応用され,がん患者さんの笑顔につながるような研究をしたいと考えている.
ウェブサイトhttps://www.jfcr.or.jp/chemotherapy/department/fundamental/index.html
趣味走ること,サッカー,自転車.