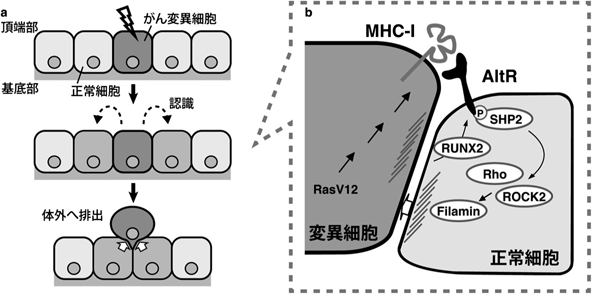
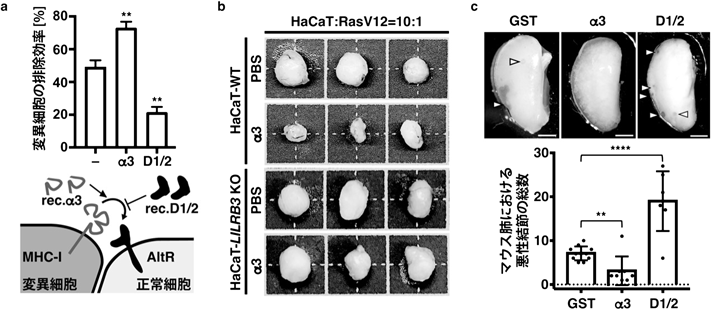
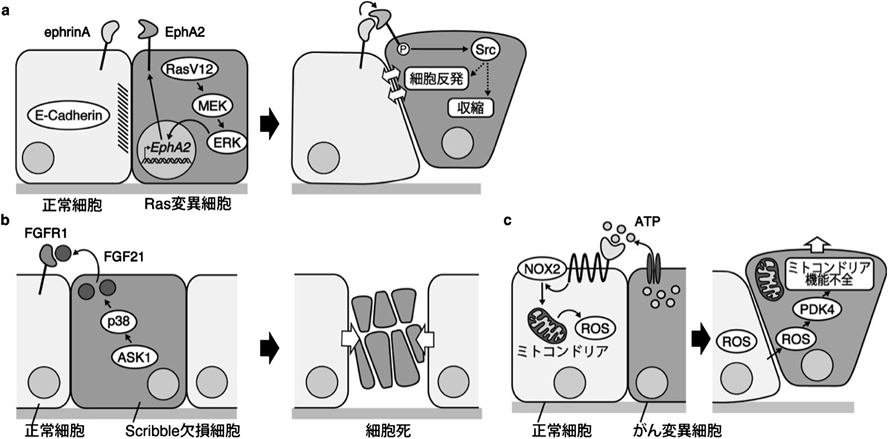
哺乳類上皮細胞によるがん変異細胞の認識と排除Mammalian epithelial cells recognize and eliminate transformed cells
東京薬科大学生命科学部生命医科学科School of Life Science, Tokyo University of Pharmacy and Life Science ◇ 〒192–0392 東京都八王子市堀之内1432–1 ◇ 1432–1 Horinouchi, Hachioji-shi, Tokyo 192–0392 Japan
発行日:2025年2月25日Published: February 25, 2025