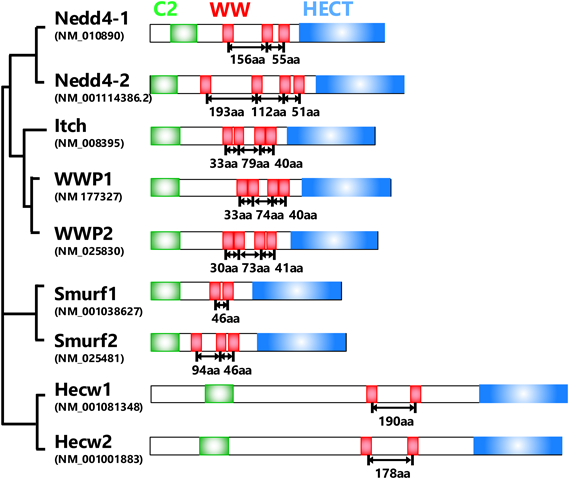
E3は,そのドメイン構造と作用機序から大きく三つのグループに分類できる.Ring Finger型,RING-between-RING(RBR)型,そしてhomologous to the E6-AP carboxyl terminus(HECT)型である.筆者らが注目しているのはHECT型に属するNedd4ファミリーE3である(図1).このファミリーの遺伝子にコードされているタンパク質はN末端のC2領域,基質タンパク質を認識する複数のWW領域,そしてC末端の酵素活性中心であるHECT領域から構成されている.酵母ではNedd4ファミリーE3が1遺伝子(Rsp5)あり,線虫では3遺伝子(wwp-1, eel-1/huwe1, Ce01588),ショウジョウバエには3遺伝子(dNEDD4, dSmurf, Suppressor of delta),哺乳類には9遺伝子がある(図1)ことからも進化の過程でNedd4ファミリーE3が増幅されてきたと考えられる4).
Nedd4ファミリーE3はそのWW領域を介して基質タンパク質のPPXYかLPXYモチーフ(以下PYモチーフ)を認識する.単一のWW領域とPYモチーフの間の結合のKdはµMのオーダーであることから,この結合は弱いといえる5).複数の連結したWW領域を介して複数の連結したPYモチーフと結合することでKdがnMのオーダーまで低下することから6),Nedd4ファミリーE3リガーゼも複数の連結したWW領域を介して複数の連結したPYモチーフを持つ基質タンパク質を認識する可能性が考えられる.大変興味深いことに,Nedd4ファミリーE3リガーゼのWW領域間の間隔はファミリーメンバーごとに多様である(図1).多様なWW領域間の間隔によってそれぞれのNedd4ファミリーのE3リガーゼが固有の基質タンパク質を認識しうる.進化の過程でさまざまなWW領域間の距離のE3リガーゼの遺伝子を増幅することで,Nedd4ファミリーE3が多様な機能を獲得したと考えられる.
このようにNedd4ファミリーE3が興味深い構造を持つ一方で,筆者が一連の研究を始めた当時,哺乳類のNedd4ファミリーE3の機能,特に個体レベルでの機能はほとんど報告されていなかった.そこで筆者はNedd4ファミリーE3に属する複数のE3リガーゼの遺伝子のコンディショナルノックアウトマウス(cKO)を作製してこれらのE3リガーゼの機能を研究することにした.
これらのE3遺伝子が発達時期の脳に多く発現していることから,神経細胞発達におけるこれらのE3の機能に注目して研究することにした.
3. 神経ネットワークの形成におけるユビキチン化研究の歴史
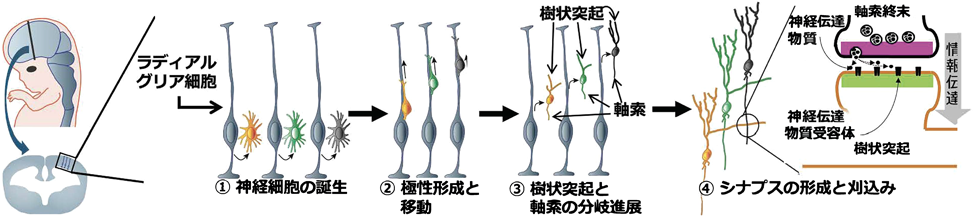
哺乳類の神経ネットワークの多くの部分を構成する興奮性神経細胞は四つのステップを経て発達する.①神経細胞の誕生(神経新生),②極性形成と移動,③樹状突起と軸索の分岐進展,④シナプスの形成と刈込みである(図2)7).神経細胞発達の異常が神経ネットワーク形成の異常につながり,自閉スペクトラム症(ASD)や統合失調症,または,てんかんなどさまざまな精神神経疾患の発症につながる.これらの発達過程の多くは線虫やショウジョウバエでも観察されることから,これらのモデル動物を使った研究が先行した.
1)線虫とショウジョウバエを使った研究
線虫とショウジョウバエを使うことによって順遺伝学(forward genetics)が可能になる.JinのグループとNonetのグループが独立にシナプス小胞がシナプスにリクルートされなくなる線虫の変異体をスクリーニングして,その原因遺伝子としてRing Finger型E3をコードするrpm-1を同定した8, 9).同時期にGoodmanのグループがショウジョウバエの神経筋接合部の形態異常を示す変異の原因遺伝子としてHighwireを同定した10).rpm-1とHighwireがオルソログであったことから種を超えてこれらのRing Finger型E3がシナプス形成に重要な役割を果たすと考えられた.独立した3グループからのこれらの研究成果は2000年の同じ号のNeuron誌に総説とともに掲載されて大きな話題になった11).
線虫を使った研究はさらに進み,Ring Finger型E3複合体であるSCF複合体とAPC複合体の構成タンパク質をコードする遺伝子fsn-1やcdc-27,またはNedd4ファミリーE3遺伝子であるwwp-1を欠損させることでrpm-1欠損変異体と同様の表現型を示すことが報告された12–14).さらに,SCF複合体の別の構成タンパク質であるSKR-1とSEL-10のユニークな機能が報告された15).skr-1欠損変異体とsel-10欠損変異体の両者でシナプス刈込みの減少が観察されたことから,このSCF複合体がシナプス刈込みを促進する.SKR-1がシナプス結合を形成する膜貫通タンパク質SYG-1の細胞内領域に結合することで,SCF複合体の形成が阻害される.その結果SYG-1が濃縮したシナプスではシナプス刈込みが起きないことが明らかにされた.SYG-1が濃縮した特定のシナプスをシナプス刈込みから守るために,SCF複合体が本質的な役割を果たすことを示す興味深い結果といえる.
線虫とショウジョウバエを使った順遺伝学研究が大きなブレークスルーをもたらしたかのようにみえたが,2010年ごろからこれらのモデルを使った研究の発展が滞っている.その理由として,rpm-1/HighwireのオルソログであるPhr1/MYCBP2の欠損マウスや欠損患者が線虫やショウジョウバエとは異なる表現型を示すことと16, 17),線虫とショウジョウバエを使った研究では神経筋接合部シナプスを観察している一方で哺乳類では主に中枢神経系シナプスを研究対象としているということ,そして,シナプス微細形態とシナプス構成タンパク質の構造がショウジョウバエと哺乳類では異なるということがある18–20).
特異的ユビキチン化の神経ネットワークの形成における役割を研究するためには哺乳類のモデル動物を使うことが重要であるといえる.
2)遺伝子欠損マウスを使った研究
初代培養神経細胞が構成する神経ネットワークを活性化することによってシナプス構成タンパク質がユビキチン化されることが古くに報告されていた21).また,プロテアソーム阻害薬によってシナプスレベルでの学習と記憶の基礎と考えられている長期増強(LTP)が抑制されることも報告されていた22).一方,特異的ユビキチン化が神経ネットワークの発達と機能をどのように制御しているかについての研究は遅れていた.唯一例外的に研究されてきたのが,HECT型E3をコードするUbe3a遺伝子の機能である.もともとUbe3aはヒトパピローマウイルス(HPV)のE6タンパク質に結合するタンパク質をコードする遺伝子E6-APとして同定,クローニングされた23).クローニングの2年後,この遺伝子の産物が哺乳類のタンパク質として初めてE3活性を持つタンパク質であることが報告された24).ヒトのUBE3Aの欠損が,精神遅滞を伴う発達障害であるAngelman症候群を引き起こすことが報告されて25, 26),さらにUbe3a欠損マウスがAngelman症候群様の表現型を示すことが報告されて以来27),この遺伝子の研究が加速した.また,大変興味深いことにUBE3Aの重複によってASDが引き起こされることが明らかになり28),Ube3aの研究は2000年ごろに俄に活発になった.
しかし,E3活性を持たないUbe3aをマウスのゲノムに重複させた場合でもASD様の表現型を示すことから29, 30),Ube3aはE3としての役割以外の役割を果たすことでASD発症に関与している可能性が考えられる.また,当初はシナプス後部でのUbe3aの機能が破綻することがAngelman症候群を引き起こすと考えられていたが31),現在は,核内,あるいはシナプス前部でのUbe3aの機能の欠損がAngelman症候群の病態に重要であると考えられており,その発症の機序はいまだに不明な点が多い32, 33).
筆者らは,Ube3aと同じHECT型E3であるNedd4ファミリーE3が発達過程の脳に多く発現していることに注目して,Nedd4ファミリーの各メンバーがそれぞれに固有の機能を持つことで複雑な神経ネットワーク形成を制御しているとの仮説のもとに各メンバーの機能の研究を始めた.
1)Nedd4-1(NEDD4)
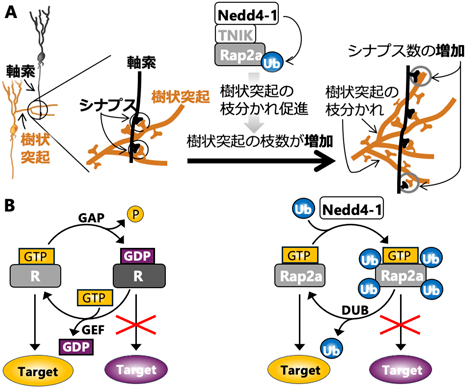
Nedd4-1はNeuronal precursor expressed developmentally downregulated geneの一つとして報告された遺伝子である34).そのmRNAの発現が胎生期の脳に多いことから,神経細胞の誕生(神経新生)に重要な遺伝子であると予想されていた.実際に,神経新生に重要な役割を果たすことが神経堤細胞についてではあるが報告されている35).そのmRNA発現は生後に減少するものの,加齢マウスの脳にも多くのNedd4-1 mRNAが発現していることから神経新生以外にも重要な機能を持つと考えられる.筆者らは,神経細胞特異的条件つきNedd4-1欠損マウス(Nedd4-1 ncKO)を作製してNedd4-1の神経新生後の神経ネットワーク形成における役割を解析したところ,この遺伝子が樹状突起の分岐と進展を促進することを見いだした(図3A)36).この現象の分子機序を理解する目的でNedd4-1の基質タンパク質認識領域に結合するタンパク質を生化学的に精製して質量分析で同定した.その結果,セリン・トレオニンキナーゼであるTRAF2/Nck-interacting kinase(TNIK)を同定した.TNIKは低分子量GTPaseであるRap2aの標的タンパク質であり,TNIKまたはRap2a活性型変異体を神経細胞に過剰発現すると樹状突起の分岐と進展が抑制される.筆者らは,Nedd4-1-TNIK-Rap2aが三者複合体を形成することと,Rap2aがNedd4-1-TNIK-Rap2a複合体形成に依存してNedd4-1によってユビキチン化されることを見いだした(図3A).興味深いことにRap2aはNedd4-1によってモノユビキチン化,またはダイユビキチン化を受ける.このユビキチン化を受けたRap2aは標的タンパク質と結合できなくなることが明らかになった.Rap2aユビキチン化がRap2aの機能を阻害することで,樹状突起の分岐・進展を促進して,結果としてシナプス数を増加させると結論した(図3A).
Rasファミリーのモノユビキチン化が細胞内局在の制御に重要であることは報告されていたが37),ユビキチン化による機能阻害は,低分子量GTPaseの制御としては新しいものであり,低分子量GTPaseの中でもRap2aの活性制御に特徴的なものかもしれない(図3B).Rap2aはほかの低分子量GTPaseと異なり,細胞内でのGTPase活性が弱い.そのため,細胞内の半数以上のRap2aがGTP結合型として存在する38).これらの結果から,筆者らはユビキチン化がGTP結合型Rap2aの活性を決定する新しい機序であり(図3B),ユビキチン化によるRap2aの活性制御が哺乳類神経細胞の樹状突起の分岐進展制御に重要であるという結論に至った.後の研究で,グリオーマ細胞でのRap2aユビキチン化がRap2aの活性制御に重要であることと,乳がん細胞でのRap2aユビキチン化がRap2aの細胞内局在制御に重要であることが,他のグループから報告された.Rap2aユビキチン化が多様な役割を果たす可能性が考えられる39, 40).
この成果を発表した筆者らの論文とback-to-backでアフリカツメガエルの視神経の軸索の進展がNedd4-1によって制御されているという内容の論文が発表された41).Nedd4-1がPTENをユビキチン化することでがん原遺伝子として機能するという先行論文のデータ42, 43)に基づいて,アフリカツメガエルの視神経細胞でNedd4-1がPTENをユビキチン化することが軸索の進展を促進すると報告している.しかし,ひな形になっているNedd4-1とPTENの関係を示した2報の論文42, 43)についてその結果の解釈に問題がある可能性が指摘されている44).さらに,哺乳類神経細胞では,Nedd4-1がPTENをユビキチン化しているのではなく,PTENがmTOR経路を介してNedd4-1の発現を抑えることで樹状突起の分岐進展を抑制していることを筆者らは明らかにした45).
この一連の研究は哺乳類のNedd4-1の機能を組織・個体レベルで明らかにした先駆け的研究となっており,これに続いて,ここ10年間ほどでNedd4-1の機能に関しての論文が多く発表されてきた.急性血中酸素濃度低下を感知するTRPA1チャネル46)やalpha-synuclein47, 48),RTP80149),FGFR50)など,思いもよらなかった基質タンパク質が次々と同定されてきた.
Nedd4-1が多くの基質タンパク質をユビキチン化することとNedd4-1を欠損させると胎生致死となることからも,Nedd4-1は生体の発達と維持に非常に重要な遺伝子であると考えられる.Nedd4-1の酵素活性中心であるHECT領域を標的にした薬剤は重大な副作用を引き起こすことが予想される.Nedd4-1に関する基礎研究の成果を創薬研究に発展させるためには,Nedd4-1・基質タンパク質複合体の立体構造をもとにNedd4-1・基質タンパク質結合の特異的阻害剤を開発する必要がある.
2)Nedd4-2(NEDD4L)
Nedd4-2は,基質タンパク質を認識するWW領域が一つ多いこと以外はNedd4-1と非常に相同性が高い(図1).そのためか,in vitroユビキチン化アッセイでは一部の基質タンパク質はNedd4-1とNedd4-2の両者によって認識されてユビキチン化される51, 52).しかし,in vivoではNedd4-2の基質タンパク質のなかでも重要なものはNedd4-1によるユビキチン化を受けていないようである53, 54).たとえば,Connexin43とKir4.1といったNedd4-2の基質タンパク質は,Nedd4-2 cKOではタンパク質発現量が増加していたが,Nedd4-1 cKOでは増加していなかった54).これらのin vivoでの研究結果とin vitroユビキチン化アッセイの結果の食い違いは,Nedd4-1とNedd4-2の細胞内局在の差によって説明できる.Nedd4-1が細胞質に一様に分布している一方でNedd4-2は細胞膜に濃縮している.そのため,in vivoでは,Nedd4-2はNedd4-1に比べてチャネルタンパク質のように細胞膜に濃縮しているタンパク質をユビキチン化する傾向があると考えられる.チャネルタンパク質をコードする遺伝子の異常は,QT延長症候群や,嚢胞性線維症,Bartter症候群,Liddle症候群などさまざまな疾患を引き起こす55, 56).実際に,Nedd4-2によってユビキチン化されるチャネルタンパク質の多くが疾患原因遺伝子の産物である(表1)57–65).なかでもてんかん原因遺伝子にコードされているタンパク質がNedd4-2の基質タンパク質になっていることが多い.
表1 Nedd4-2基質をコードする遺伝子とその変異が引き起こす疾患| Nedd4-2基質タンパク質名 | 遺伝子名 | 遺伝子変異による疾患名 | 結合・ユビキチン化に関する文献 |
|---|
| ENaC-αサブユニット | SCNN1A | 高血圧(Liddle症候群) | Kamynina, et al., FASEBJ, 200165) |
| ENaC-βサブユニット | SCNN1B | 高血圧(Liddle症候群) | Kamynina, et al., FASEBJ, 200165) |
| ENaC-γサブユニット | SCNN1G | 高血圧(Liddle症候群) | Kamynina, et al., FASEBJ, 200165) |
| Connexin43 | GJA1 | 不整脈 | Altas, et al., JCB, 202454) |
| Kir4.1 | KCNJ10 | てんかん(EAST/SeSAME症候群) | Altas, et al., JCB, 202454) |
| Nav1.2 | SCN2a | てんかん(Dravet症候群),ASD | Fotia et al., JBC, 200464) |
| Nav1.5 | SCN5a | 不整脈(Brugada症候群) | van Bemmelen et al., Circulation Research, 200463) |
| Nav1.7 | SCN9a | 先天性無痛無汗症 | Fotia et al., JBC, 200464), Laedermann et al., JCI, 201361) |
| Kv7.1 | KCNQ1 | QT延長症候群 | Jespersen et al., Cardiovascular Research, 200761) |
| Kv7.2 | KCNQ2 | てんかん(West症候群) | Ekberg, et al., JBC, 200760) |
| Kv7.3 | KCNQ3 | てんかん,ASD | Ekberg, et al., JBC, 200760) |
| ΔF508-CFTR | CFTR | 嚢胞性線維症 | Caohuy et al., JBC, 200959) |
| TTYH2 (Tweety2) | TTYH2 (Tweety2) | 報告なし | He et al., JBC, 200858) |
| TTYH3 (Tweety3) | TTYH3 (Tweety3) | 報告なし | He et al., JBC, 200858) |
| ATA2 | ATA2 | 報告なし | Hatanaka et al., JBC, 200657) |
てんかんはチャネル病と呼ばれるように,てんかん原因遺伝子の多くがチャネルタンパク質をコードする.このようなチャネルタンパク質は,興奮性シナプス後部,抑制性シナプス後部,樹状突起,軸索起始部,軸索,そしてシナプス前部アクティブゾーン(AZ)に局在している.細胞膜局所でイオン流入を介して神経細胞の興奮性を制御している.一方で,アストロサイトのチャネルタンパク質は細胞外イオン濃度の恒常性を維持することで神経細胞の興奮性を安定させている.アストロサイトでのチャネルタンパク質遺伝子の欠損が細胞外イオン濃度の上昇を引き起こし,その結果,神経細胞の興奮性の上昇,神経ネットワークの過剰な活性化,そして,てんかん発症につながると考えられる.しかし,てんかん病態における神経ネットワークの過剰な活性化の機序は不明な点が多い.
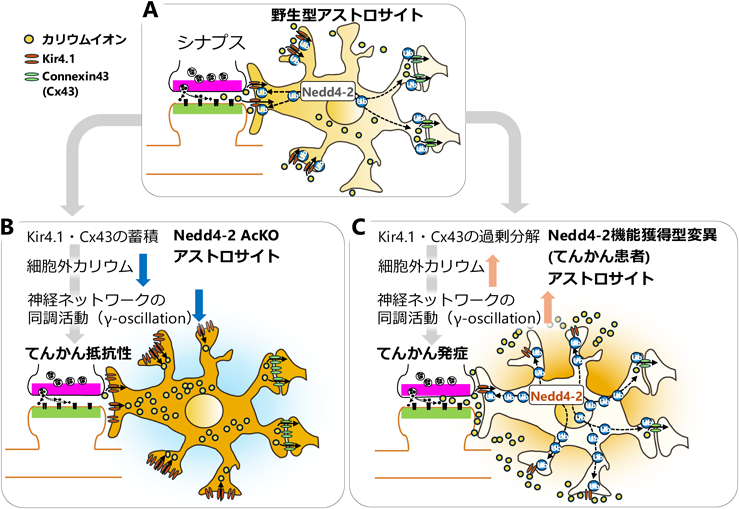
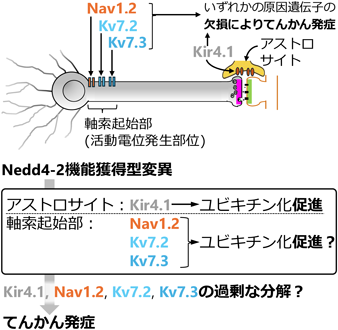
複数のてんかん患者でのNedd4-2遺伝子変異が報告されている66, 67).これについていくつかの論文が発表されたが,Nedd4-2がどのようにてんかん発症に関与しているのかについて不明な点が多くあった.筆者らは,Nedd4-2の基質タンパク質として,てんかん原因遺伝子産物でアストロサイトに多く発現するKir4.1とアストロサイトに多く発現するギャップ結合構成タンパク質Connexin43を同定した(図4A).これらの基質タンパク質がNedd4-2によってユビキチン化されることでリソソームでの分解に導かれることを見いだした.Kir4.1とConnexin43はカリウム透過性が高い(図4A).細胞外カリウムがKir4.1を通過してアストロサイト細胞内に流入してConnexin43を介して隣接するアストロサイトに拡散する.Kir4.1とConnexin43は,いずれも細胞外カリウム濃度を一定の濃度以下に保つのに重要なタンパク質である68).細胞外カリウム濃度が増加すると,細胞外陽イオンが増加することにより細胞外の主な陽イオンであるナトリウムが細胞内に流入しやすくなる.このように,神経細胞と神経ネットワークは興奮しやすくなる.細胞外カリウム濃度を一定に保つことは神経ネットワーク同調活動(γ-oscillation)を維持するのに非常に重要である.実際に,アストロサイト特異的Nedd4-2 cKO(Nedd4-2 AcKO)では,Kir4.1とConnexin43が分解されないために発現量が増加し,その結果,神経ネットワーク同調活動(γ-oscillation)が低下することを見いだした(図4B).神経ネットワーク同調活動(γ-oscillation)の過剰な活性化がてんかん発作につながると考えられていることから,Nedd4-2 AcKOはてんかん抵抗性を獲得したといえる.これは,てんかん患者でNedd4-2変異が見つかったことと一見矛盾した結果である.興味深いことにてんかん患者に見つかったNedd4-2の変異はすべて機能獲得型変異であることを筆者らは見いだした.こうした変異を持つ患者のアストロサイトではKir4.1とConnexin43のユビキチン化と分解が亢進してこれらの基質タンパク質の発現量が低下すると考えられる.その結果,細胞外カリウム濃度が増加するために,神経ネットワーク同調活動が過剰に活性化しててんかんを発症するという作業仮説を筆者らは提案している(図4C)54).
神経細胞の軸索起始部においてもNedd4-2はてんかん原因遺伝子産物をユビキチン化している(表1,図5).Nedd4-2の機能獲得型変異を持つてんかん患者では,神経細胞でこれらの基質タンパク質のユビキチン化が亢進して過剰に分解される可能性がある.これにより,てんかん原因遺伝子欠損と同様の機序でてんかんにつながる可能性が考えられる.Nedd4-2によるユビキチン化を阻害することで効果的な抗てんかん薬創薬につながる可能性がある.実際に,これらのてんかん原因遺伝子産物に対するナノボディーを脱ユビキチン化酵素に融合させたタンパク質を発現させることで治療に結びつける創薬研究が始まっている69).
3)Wwp1(WWP1)とWwp2(WWP2)
哺乳類ではWwp1, Wwp2,そしてItchyの3種類のE3遺伝子が高い相同性を示す(図1).Itchyはその名のとおり,かゆみの異常亢進を示す自然発生変異マウスの原因遺伝子として同定された70).その後の研究で,ItchyがコードするItchはJUNBをユビキチン化することでT細胞の分化を制御するE3であることが明らかになった71).免疫系でのItchの機能についての研究が進んだ一方で,哺乳類のWwp1とWwp2の研究は近年まで盛んではなかった.
哺乳類のWwp1とWwp2の遺伝子は1997年にクローニングされ,生化学的に解析されてきた72).しかし,その個体レベルでの機能についてはまたしても線虫を使った研究に先行された.線虫ではwwp-1のみ発現しているが,この遺伝子が一躍有名になったのはTony Hunterのグループからの論文によるところが大きい73).この論文ではwwp-1を過剰発現することで線虫の寿命が20%も延長したと報告されている.寿命延長は食事制限で達成されることがすでに知られていたが,食事制限の下流でWWP-1を介したユビキチン化が機能していることが明らかにされた.後の研究で,食事制限の下流でWWP-1が転写制御因子であるKrüppel-like factor(KLF-1)をモノユビキチン化することでKLF-1の機能を促進していることが報告された74).
哺乳類ではがん研究の分野でWwp1とWwp2の研究が進んできた.タンパク質またはmRNAのレベルで,Wwp1の発現量が前立腺がん75)と乳がん76)で増加しており,Wwp2の発現量は前立腺がん77),口腔がん78),肝細胞がん79),肺腺がん80)で増加していた.特にWwp2発現量は口腔がんと肝細胞がんの予後と密接な関係があり,これらのがん由来の培養細胞の細胞分裂の速度がWwp2のノックダウンによって抑えられる79).このようにWwp1とWwp2のがん遺伝子としての研究はかなり進んできた.
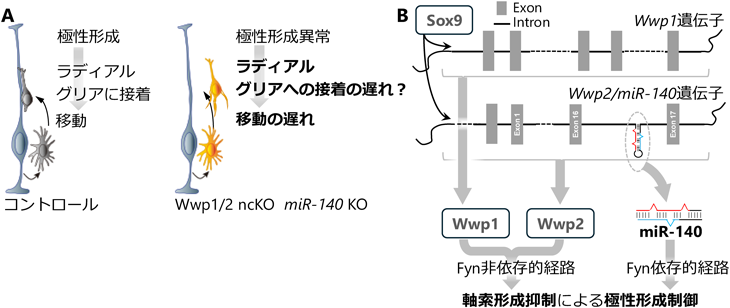
一方で,Wwp1, Wwp2ともに分裂しない神経細胞で発現量が多いにもかかわらず,哺乳類神経細胞での役割はいまだに十分に明らかになっていない.筆者らはWwp1とWwp2の相同性が非常に高いことからこれらが共通の機能を持っていると仮定した(図1).神経細胞でのこれらのE3の機能を研究する目的でWwp1f/f;Wwp2f/fマウスに神経細胞特異的にCreを発現させた81).以降このマウスをWwp1/2 ncKOと呼ぶ.Wwp1とWwp2の発現量が胎生中期に多いためにWwp1/2 ncKOを使って神経細胞の極性形成と移動について解析を行った.その結果,コントロール神経細胞は1本の軸索を伸ばすが,Wwp1/2 ncKO神経細胞は複数の軸索を伸ばしていたことから,極性形成に異常があると結論した.さらに,細胞移動に遅れが生じていた.これらのE3が協調して神経細胞の極性形成を正常化させて移動をスムーズにさせていると結論した(図6A).
大変興味深いことに,Wwp2遺伝子のイントロン領域にmiR-140がコードされている(図6B).この遺伝子構造から,共通の転写制御因子によってWwp2のpre-mRNAの転写が活性化されてWwp2とmiR-140がほぼ同時に神経細胞内に発現して協調して機能するとの仮説を立てた.この仮説を検証したところ,筆者らは,miR-140 KO神経細胞がWwp1/2 ncKO神経細胞と同様に複数の軸索を進展させることと遅れた細胞移動を示すことを明らかにした.さらに,miR-140が,Wwp2とは別のシグナル伝達経路を介して極性形成を正常化させて移動をスムーズにさせていることを報告した(図6B)81).Sox9が共通の転写制御因子としてこれらの遺伝子発現を制御していることも見いだした.イントロンにコードされているmiRNAがタンパク質をコードするホストmRNAと同じ機能を持つことを証明した画期的な論文といえる.この発見は同じ号のNeuron誌に特集されるなど注目を集めた82).
筆者らの一連の研究により,個々のNedd4ファミリーE3が神経細胞の発達と機能の制御で各々独自の役割を果たすことが明らかになった.進化の過程で増加したNedd4ファミリーE3遺伝子の多様な役割によって,複雑化した哺乳類神経細胞の発達と機能が制御されている可能性が考えられる.
6. 今後の展望:E3遺伝子の変異が原因となる発達障害の病態解明にむけて
4節で説明したように,特異的ユビキチン化による神経細胞の発達と機能の制御機序が近年徐々に明らかになってきた.一方で,5節であげたE3遺伝子の変異が原因となる疾患の発症機序は十分には明らかになっていない.さらに,Ring Finger型E3をコードしている発達障害原因遺伝子も数多く報告されている.これらの変異がどのように発達障害発症につながるかを理解するためには,それぞれのE3の基質タンパク質を同定する必要がある.そしてE3遺伝子欠損マウスの脳で基質タンパク質がユビキチン化されないことがどのような機序で発達障害発症につながるかを,生化学,形態学,電気生理学,行動学などの手法を組み合わせて明らかにするというアプローチが最も堅実である.
E3の基質タンパク質を同定する手法として,リコンビナントE3タンパク質に結合するタンパク質のスクリーニング36)やタンパク質マイクロアレイを使ったユビキチン化基質スクリーニング51, 91)など,in vitroのスクリーニング法がとられてきた.最近では,E3遺伝子を欠損したマウスや培養細胞を使った定量的質量分析によるスクリーニングが主流になりつつある.E3遺伝子欠損により発現量が増加したタンパク質やユビキチン化レベルが減少したタンパク質をスクリーニングする.これらのin vivoのスクリーニング法によってin vitroのスクリーニング法よりもより高い信頼性で内在性E3の基質を同定できる54, 92).筆者らは,Ube3B欠損マウスで発現量が増加したタンパク質をスクリーニングすることで脱リン酸化酵素であるカルシニューリンの触媒サブユニットPpp3ccをUbe3Bの基質タンパク質として同定して,このユビキチン化が樹状突起の成長を促進してシナプス形成を抑制することを見いだした93).
ユビキチン化による神経細胞の発達と機能の制御機序の研究は,リン酸化や転写・翻訳による制御機序の研究に比べて進んでいない.5節で説明したようにE3をコードする疾患原因遺伝子が多いにもかかわらず未解明な部分が多いのが現状である.その理由として,ユビキチン化は,基質タンパク質の発現量だけでなく空間的ダイナミクスを制御するなど,複雑な役割を果たしていることがあげられる.
特異的ユビキチン化が基質タンパク質の空間的ダイナミクスにどのように影響するのかを,ライブイメージングや超解像蛍光顕微鏡を駆使して研究することが今後の研究で必要になると考えられる.筆者らも近年の普及よりかなり早い段階から,20 nm程度の実質分解能の超解像蛍光顕微鏡技術を取り入れて独自に発展させ,それらを駆使して研究を進めている(図7).このような顕微鏡技術を駆使することで神経科学領域だけでなく,広い領域でのユビキチン化研究に貢献したい.