1. オートファジー研究の発展
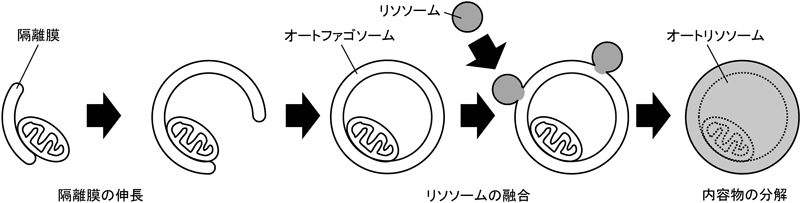
マクロオートファジー(以下オートファジー)は真核生物に広く保存されている細胞内の分解システムである1, 2).この過程では,隔離膜(またはファゴフォア)と呼ばれる膜構造が伸長し,オートファゴソームと呼ばれる二重膜で細胞質成分を取り囲む.次いで,オートファゴソームがリソソームと融合することで,囲まれていたタンパク質やオルガネラが分解される(図1).オートファジーはタンパク質やオルガネラの品質管理による細胞内の恒常性維持や飢餓適応などにおいて重要な役割を担っている.
大隅良典先生が出芽酵母でオートファジー関連遺伝子を発見3)したのをきっかけに分子生物学的な研究が始まったオートファジーは,その哺乳類ホモログの同定,およびマウス個体における生理的重要性の発見に至り,さらに世の中の注目を浴びることとなった.筆者がオートファジーの研究を始めた2007年の時点ではPubMedで検索しても論文が「全部で」2000件程度といったところだったが,今や10万件近い論文を擁する一大研究分野となっている.筆者が2016年までに携わってきた研究にはオートファジーの分子機構,上流制御因子,定量法,ノックアウトマウス,制御薬の探索などがあるが,いずれのトピックもその後大きく発展している.本稿では,筆者が携わってきた研究を含めながらそれぞれのトピックを解説し,オートファジーに関する現在の全体像を概観することとしたい.なお,ミクロオートファジーおよびシャペロン介在性オートファジーについては本稿では取り扱わない.また,本稿では特に断りのない限りは哺乳類のオートファジーについて記す.
2. オートファジーを駆動するタンパク質群
1)ATGタンパク質群の遺伝学的ヒエラルキー
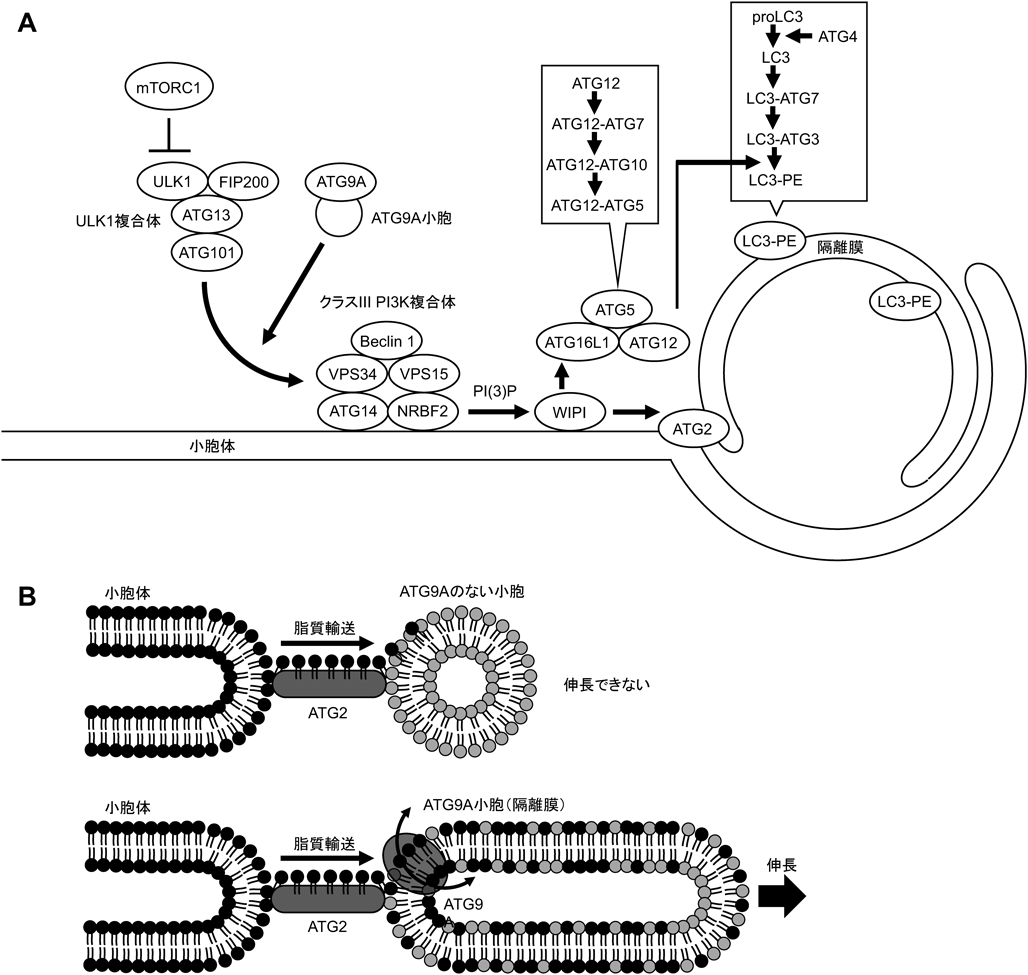
オートファゴソームの形成には一連のAtg(autophagy-related)に遺伝子にコードされるATGタンパク質群をはじめとする多数のタンパク質が必要となる2, 4–10).哺乳類の主要なATGタンパク質群は図2Aに示したように複数のグループに分類することができる.ULK複合体,ATG9A,クラスIII PI3K複合体,WIPI, ATG12–ATG5–ATG16L1複合体,およびLC3/GABARAPである.これらのタンパク質群は,オートファゴソームの形成において重要な役割を担っている.多くのタンパク質があり複雑にみえるが,これらATGタンパク質群は遺伝学的ヒエラルキーの概念により整理することができる.ATG分子群はオートファゴソームが形成される部位に集積することで,蛍光顕微鏡で輝点として観察される.大隅研究室の鈴木らは各Atg遺伝子が欠損した出芽酵母におけるほかのAtgタンパク質の輝点形成を網羅的に分析することで,Atgタンパク質の集積における遺伝学的な上下関係を明らかにした11).その後,同様のヒエラルキーが哺乳類細胞においても成立することが示された12).一番上流に位置するのはULK複合体とATG9A,次がATG14を含むクラスIII PI3K複合体,その下にWIPI, ATG12–ATG5–ATG16L1,最後にLC3/GABARAPという形になっている(図2A).
2)ULK複合体
ULK複合体はULK1(unc-51 like kinase 1)またはULK2, FIP200(focal adhesion kinase-family-interacting protein of 200 kDa,別名RB1CC1),ATG13, ATG101から形成される4, 5).ULK1, ULK2は酵母Atg1の,FIP200は酵母Atg17のホモログにあたる2).酵母Atg1複合体およびULK複合体は液-液相分離によってオートファゴソーム形成部位へと集積する13, 14).ULK複合体が遺伝学的ヒエラルキーの「最上流」に位置していることは,FIP200の欠損細胞で下流のATG分子群のオートファゴソーム形成部位への集積がみられなくなることにより示された12)さらに筆者はATG13欠損細胞,ATG101欠損細胞において同様の表現型を確認している15, 16).ただしULK1の欠損によるオートファジーへの影響は限定的であり,ULK1/2両方を欠損した細胞でも若干のオートファジー活性が検出されることからULK1/2はオートファジーに必須とまではいえないと考えられる17, 18).この結論は,ULK1結合部位を欠損するATG13の発現がATG13欠損細胞のオートファジーを部分的に回復させられることからも支持されている19).
ATG101は下流ATG分子群のリクルートにおいて重要な役割を担っている.出芽酵母にホモログが存在しないATGタンパク質ということでこのように命名されたATG101は,ATG13を介してULK複合体に組み込まれている16, 20, 21).ATG13に結合できないATG101変異体がオートファゴソーム形成部位に局在できないこと,およびATG101欠損細胞でもほかのULK複合体構成因子がオートファゴソーム形成部位に局在できることから,ATG101はULK複合体の内部では「下流」に位置するタンパク質だと考えられる16).ATG101は,トリプトファン(W)とフェニルアラニン(F)が露出した特徴的な構造「WFフィンガー」を持っている.WFフィンガーに変異を持つATG101変異体はATG13に結合でき,他のULK複合体構成因子とともにオートファゴソーム形成部位へと集積するが,このATG101変異体を発現する細胞では下流のATG分子群の集積はみられなくなる16).さらにその後,ATG101のC末端領域がATG14と結合することでPI3K複合体をリクルートするという報告22),およびATG13–ATG101複合体がATG9Aに結合することでATG9小胞をリクルートするという報告23, 24)がなされている.しかしながら,これらの報告におけるATG14結合部位およびATG9A結合部位は,いずれもWFフィンガーとは異なっているため,WFフィンガーが具体的に何をしているのかは現時点でも不明である.
オートファジーは栄養飢餓によって活性化するが,この栄養に応じたオートファジーの制御に重要な役割を果たしている因子がmTOR(mechanistic target of rapamycin)である.mTORは構成因子の異なる少なくとも2種類の複合体を形成するが,これらのうちmTOR complex 1(mTORC1)はアミノ酸により活性化し,翻訳や細胞成長を促進し,オートファジーを抑制する役割を持っている25).筆者らはmTORC1がULK複合体に栄養依存的に会合し,ULK1およびATG13をリン酸化することを明らかにした26, 27).その後,他のグループにより具体的なリン酸化部位とそのオートファジー制御における役割も明らかになっている28, 29).
3)クラスIII PI3K複合体とWIPI
ULK複合体の下流で機能する重要なタンパク質複合体の一つがクラスIII PI3キナーゼ(phosphoinositide 3-kinase:PI3K)複合体である5).PI3KというとインスリンシグナルにおけるPI3Kが有名だが,それはPI(4,5)P2をPI(3,4,5)P3へ変換するクラスI PI3Kであり,オートファゴソーム形成において働くのはPIからPI(3)Pを産生するクラスIII PI3Kである30).クラスIII PI3Kは,酵素活性を持つVPS34(vacuolar protein sorting 34),そしてBeclin 1, VPS15を共通の構成因子として複数の複合体を形成するが,オートファゴソームの形成に寄与するのはATG14とNRBF2(nuclear receptor binding factor 2)を含む複合体である5).ATG14はPI3K複合体を小胞体上に局在させる役割を持っており,過剰発現によりオートファジーを誘導することができる31).
クラスIII PI3K複合体のオートファゴソーム形成部位への集積とPI(3)Pの産生は,PI(3)P結合タンパク質であるWIPI(WD-repeat protein interacting with phosphoinositides)のリクルートに必要となる6).WIPIファミリーにはWIPI1からWIPI4までの四つのタンパク質があり,WIPI2を欠損した細胞,またはWIPI3とWIPI4の両方を欠損した細胞ではオートファジーが顕著に抑制される32, 33).WIPI2はATG16L1に結合することで,ATG16L1の隔離膜局在に寄与する34–36).一方WIPI4はATG2と結合する33).実験条件によっては,WIPI3とATG2との結合も検出できる37)ことから,WIPI3とWIPI4の両方がATG2のリクルートに関与しているものと考えられる.
4)ATG2, ATG9, VMP1,およびTMEM41B
近年,構造生物学とin vitroの実験を駆使した一連の研究により,ATG2とATG9が脂質分子の輸送により隔離膜の形成・伸長を行っていることが明らかになった38).ATG2はN末端側,C末端側それぞれで脂質膜に結合する棒状の構造をとっている37, 39).そして,酵母Atg2および哺乳類ATG2は二つの膜構造の間で脂質分子を輸送する機能を持っていることが示された40–42).これらのことから,ATG2は小胞体から隔離膜へと脂質分子を輸送することで,隔離膜の伸長に寄与しているのではないかと考えられる.また,酵母Atg9および哺乳類ATG9Aはそれぞれ三量体構造を形成し,リン脂質のスクランブラーゼとして機能することが明らかとなった43–45).ATG2による脂質輸送だけでは外側リーフレットに脂質分子が蓄積して不均衡が生じるため,脂質輸送に支障を来してしまうと考えられるが,ATG9がそれを解消することで,膜全体が伸長できるようにしていると考えられる(図2B).
VMP1(vacuole membrane protein 1)およびTMEM41B(transmembrane protein 41B)は,小胞体に局在し,オートファゴソーム形成に重要な役割を果たすタンパク質である8).VMP1は小胞体上のオートファゴソーム形成部位に局在していると考えられている12, 46).VMP1およびTMEM41Bは配列に相同性を有し,機能的にも重複性がある47).ATG2がVMP1およびTMEM41Bに結合することを考慮すると,これらのタンパク質はATG2を小胞体のオートファゴソーム形成部位につなぎとめる役割を持っているのかもしれない45).なお,VMP1とTMEM41Bもスクランブラーゼ活性を持つことが示されている45, 48, 49)が,この活性がオートファゴソーム形成においてどのような意義を持つのかは今のところわかっていない.
5)ATG12–ATG5–ATG16L1複合体
オートファゴソーム形成の下流のステップでは,ユビキチンと同様の反応機構により共有結合を形成する二つの「ユビキチン様ATG結合系」のタンパク質群が機能する2, 9).そのうちの一つ,ATG12結合系においては,ATG12がATG7とATG10を経た後ATG5に結合する(図2A)9).ATG12–ATG5はさらにATG16L1と複合体を形成する.
遺伝学的ヒエラルキーでいえば,ATG12–ATG5–ATG16L1複合体はULK複合体よりもずっと下流に位置している形になるが,これらの複合体はFIP200とATG16L1の相互作用を介して会合しているという知見も得られている50, 51).酵母ツーハイブリッドシステムの結果から示唆されていたFIP200とATG16L1の結合は内因性タンパク質の免疫沈降では検出できなかったが,筆者はケミカルクロスリンカーであるDSP[dithiobis(succinimidyl propionate)]を使って近接するタンパク質を架橋してから免疫沈降を行う手法により,両者の結合を検出することに成功した51).このDSP免疫沈降法を使ってさらに調べたところ,ULK1–FIP200–ATG13–ATG101とATG12–ATG5–ATG16L1がATG14やクラスIII PI3K活性の有無にかかわらず複合体を形成していることが示唆された.ULK複合体とATG12–ATG5–ATG16L1複合体が会合しているという知見は,ULK1とATG5が同時にオートファゴソーム形成部位へリクルートされるというライブイメージングの分析結果46)とも一致する.よって,ATG12–ATG5–ATG16L1複合体がオートファゴソーム形成部位へ移行することは,PI(3)Pとは無関係である可能性があるが,PI(3)PおよびWIPI2はこれらの複合体の安定した蓄積に必要であると考えられる.
ATG12–ATG5–ATG16L1複合体はATG3–LC3複合体を隔離膜へリクルートした後,そのE3活性によりLC3を膜脂質であるPE(phosphatidylethanolamine)へと結合させる36, 52).
6)LC3/GABARAP
LC3(microtubule-associated protein 1 light chain 3)およびGABARAP(gamma-aminobutyric acid receptor-associated protein)は酵母Atg8のホモログにあたる9).哺乳類はLC3サブファミリーのLC3A, LC3B, LC3CおよびGABARAPサブファミリーのGABARAP, GABARAPL1, GABARAPL2の全部で六つのAtg8ホモログを持っている.ユビキチン様ATG結合系の一つであるLC3結合系では,これらのタンパク質がまずATG4による切断を受け,露出したC末端のグリシン残基を介してATG7とATG3,次いでPEに結合することでオートファゴソーム膜へと局在する9).ここまでに述べたATG分子群は一過的にオートファゴソーム形成部位に集積し,オートファゴソームが完成する前に離散するが,LC3は完成したオートファゴソームの膜上にとどまることからオートファゴソームのマーカータンパク質として用いられる53).
ATG結合系のタンパク質群のオートファジーにおける機能および必要性については,ATG5およびATG7を欠損する細胞でもオートファジーが観察されるという報告54)があって以降,議論があったが,その後ATG3, ATG5,またはATG7を欠損した細胞ではオートファジーは完全には止まらないものの,伸長した隔離膜がオートファゴソームになりにくくなること,そしてリソソームと融合した後のオートファゴソームの内膜が分解されにくくなることがわかった55).酵母Atg8とその哺乳類ホモログについては,近年の研究で膜の伸長,形態形成,リソソームとの融合などにおいて重要な役割を果たしていることが明らかになってきている52, 56–59).
7)オートファゴソームの完成とリソソームの融合
伸長した隔離膜がオートファゴソームになる段階ではESCRT(endosomal sorting complexes required for transport)のタンパク質群が膜の閉鎖を行う60, 61).完成したオートファゴソームにはSNARE(soluble N-ethylmaleimide-sensitive-factor attachment protein receptor)であるSTX17(syntaxin 17)およびYKT6がリクルートされ,これらがリソソームとの融合に重要な役割を果たす62, 63).オートファゴソームとリソソームの結合は,HOPS(homotypic fusion and vacuole protein sorting)複合体を含む複数の結合因子によって仲介される64, 65).
8)選択的オートファジーと受容体タンパク質
オートファジーでは細胞内成分の非選択的な分解が生じるが,特定のタンパク質やオルガネラの選択的分解も行われる66, 67).最もよく知られている例はミトコンドリアの選択的な分解「マイトファジー」であろう.パーキンソン病関連因子であるParkinが損傷ミトコンドリアにリクルートされ,オートファジーによるミトコンドリアの選択的な分解を誘導するという発見は,選択的オートファジーとその分子機構の研究において重要な契機となった68).現在ではミトコンドリアのほか,凝集体,細菌,リソソーム,ペルオキシソーム,リボソーム,グリコーゲン,フェリチン,脂肪滴など,さまざまな細胞内成分が選択的オートファジーの基質となることが明らかになっている66, 67).選択的オートファジーにおいては,分解の標的となる構造体がp62(別名SQSTM1),TAX1BP1(Tax1-binding protein 1),NDP52(別名CALCOCO2),NBR1(neighbor of Brca1 gene 1),OPTN(Optineurin)といった受容体タンパク質により認識される.受容体タンパク質による基質の認識には,いくつかのケースで標的構造体のユビキチン化が重要となる66, 67).受容体タンパク質はLC3と結合することで標的構造体とオートファゴソーム膜の間を橋渡しするが,これは選択的オートファジーのメカニズムの説明としては十分ではない.近年の研究により,受容体タンパク質はULK複合体をリクルートすることで基質上から隔離膜の形成を促すことが示されている69–71).また,選択的オートファジーにおいては受容体タンパク質の液-液相分離が重要であるという知見が得られてきている72–74).隔離膜による液滴の取り囲みは液滴とオートファゴソーム膜間の「ウェッティング効果」によって推進される75).
3. オートファジーの上流制御因子mTORC1の制御機構と維持機構
1)栄養シグナルによるmTORC1の制御メカニズム
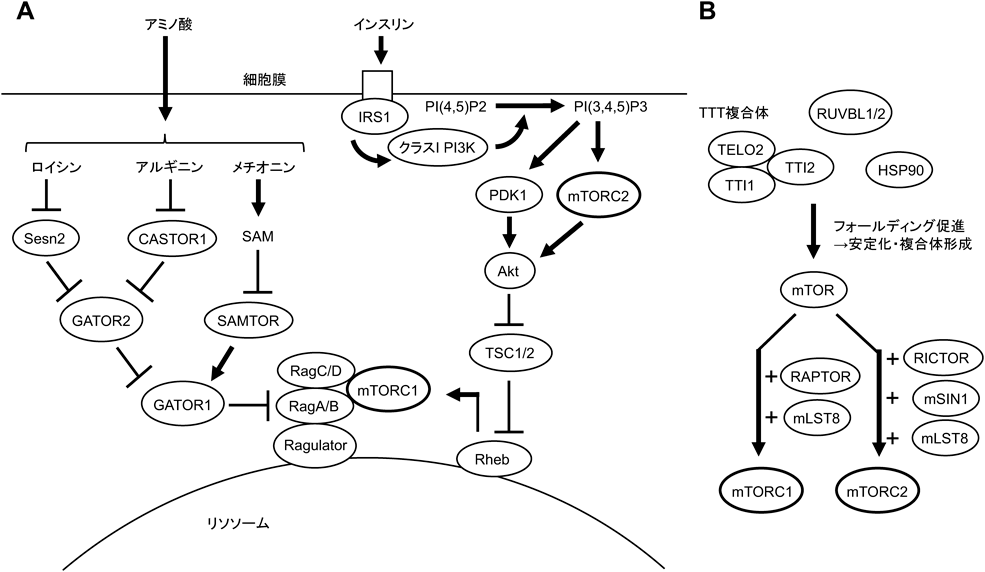
栄養飢餓はmTORC1の不活性化を介してオートファジーを誘導する.栄養に関するmTORC1の主要な上流制御シグナルとしては,インスリンなどに応答するクラスI PI3Kの経路と,アミノ酸に応答するアミノ酸経路がある25)(図3A).生体内においては,このインスリン経路とアミノ酸経路が臓器・組織によって異なる重みでmTORC1およびオートファジーを制御しており,肝臓ではアミノ酸が,筋肉ではインスリンがmTORC1およびオートファジーの制御に大きな役割を果たしている76).mTORC1シグナルの図は現在では広く受け入れられている教科書的な知識であろうが,アミノ酸経路の分子機構については,実は2008年まではほとんど何もわかっていなかった.しかしアミノ酸に応答するmTORC1上流因子Rag GTPaseの発見77)を皮切りに,マサチューセッツ工科大学の一研究チームが瞬く間に全容を解明してしまった.重要な発見をいくつか順を追ってみてみよう.Rag GTPaseはリソソーム上のRagulatorと命名されたタンパク質複合体と結合しており,mTORC1のリソソーム局在を担っていることがわかった78).次に彼らはRag GTPaseのGAP(GTPase-activating protein)としてGATOR1(GAP activity toward Rags 1)複合体,およびGATOR1の制御因子であるGATOR2複合体を同定した79).さらにGATOR2の上流制御因子として,ロイシンを感知するSestrin280, 81),アルギニンを感知するCASTOR1(cytosolic arginine sensor for mTORC1 subunit 1)82)を同定した.また,GATOR1の上流因子として,メチオニンの代謝物であるSAM(S-adenosylmethionine)を感知するSAMTOR(SAM sensor upstream of mTORC1)を同定した83).さらに彼らを含む研究グループはこれらのタンパク質複合体やその相互作用,アミノ酸感知機構の構造生物学的基盤をも示している84–88).
インスリンシグナルでは,IRS(insulin receptor substrate)を介して活性化されたPI3Kの下流でPDK1(phosphoinositide-dependent kinase-1),またはRICTORを含むmTORC2複合体がAktを活性化する25).AktはTSC1/2(tuberous sclerosis complex 1/2)を抑制し,リソソーム上でRheb(Ras homolog enriched in brain)を活性型にすることでmTORC1を活性化させる.アミノ酸シグナルはRag-Ragulatorを介してmTORC1をリソソームに局在させることでRhebとの会合を促進すると考えられている.
2)mTOR複合体の形成におけるTTT-RUVBL1/2の役割
筆者もmTORには大いに興味を持っていたため,mTOR結合タンパク質の探索と解析に取り組んでいたのだが,この研究は結論から述べるとTTT-RUVBL1/2複合体89)によるmTOR複合体形成促進機構の解明に寄与することとなった(図3B).筆者らはmTORの新規結合タンパク質として同定されたTTI1(TELO2 interacting protein 1)が,TELO2(telomere maintenance 2)とともにmTORの複合体形成および安定化に寄与していることを明らかにした90).通常,mTOR複合体の各構成因子は細胞質画分のゲルろ過クロマトグラフィーを行うと複合体と思われる高分子領域にピークが観察されるが,Tti1またはTelo2をノックダウンした細胞においてはこの高分子領域のピークが減少し,単体と思われる低分子領域に移行していた.なお,このTTI1およびTELO2については世界の複数の研究グループが注目していたようで,同じ年のうちにTELO2, TTI1, TTI2からなる「TTT複合体」がHSP90のコシャペロンとして働き,mTORを含むPIKK(phosphoinositide-3-kinase-related protein kinase)の複合体形成および安定化を助けること,RUVBL1/2(RuvB-like 1/2)と共同して働くことなどが相次いで報告された91–93).その後,これらの複合体の構造や,mTORとの結合様式も報告されている94, 95).mTOR複合体自体は栄養状態にかかわらず存在すること,mTORとTTI1の結合は栄養状態によって影響されなかったことなどから,筆者はTTI1を含むこれらのタンパク質群はmTOR複合体の「制御」というよりは,恒常的に複合体形成を促すという意味で「維持」に寄与していると考えているが,TTI1を含むタンパク質複合体が代謝ストレスによるmTOR複合体形成やmTORシグナルの調節に寄与するのではないかという結果も報告されている96, 97).近年,TTI1遺伝子の変異が神経発達障害の患者において複数発見されており,患者由来の変異にmTOR複合体形成に影響を及ぼすものがあることが報告されている98).また,TTT-RUVBL1/2複合体はmTORC1が過剰に活性化したがん細胞に対する標的としても注目されている99).
3)mTORC1以外のシグナルによるオートファジー制御
mTORC1以外によく知られているオートファジー制御因子としてはAMPK(AMP-activated protein kinase)がある.AMPKはmTORC1の制御にも関与しているが,ULK1をはじめとしたさまざまなオートファジータンパク質をリン酸化することでオートファジーを制御していることが報告されている100).またその他にも,非常に多種多様なシグナル経路がオートファジーを制御することが報告されている101, 102).実際にmTORC1の抑制を伴わないオートファジー誘導の事例がみられる103)ことからも,mTORC1を介さないオートファジー制御機構があることは確かであると考えられるが,これら各シグナル経路がどのような条件下でどれだけオートファジーの制御に寄与しているのかは,今後明らかにしていくべき課題だといえるだろう.
4. オートファジーの定量方法
1)オートファゴソームの定量とその問題点
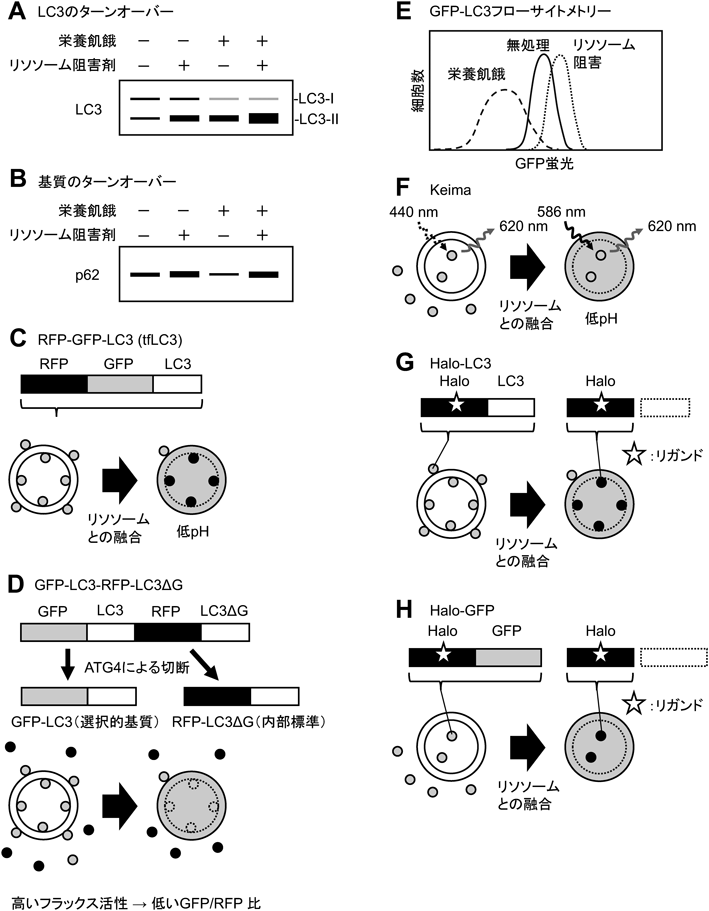
細胞や組織において,どれくらい活発にオートファジーが生じているかを定量することは,基礎研究・応用研究両面において重要な課題である.これまで,オートファジーを定量するさまざまな方法が考案されてきた53, 104).最もよく用いられてきた方法の一つは,LC3をマーカーとして検出したオートファゴソームの数をカウントする方法である.たとえばGFP-LC3を発現する細胞をアミノ酸と血清を抜いた培地で2時間培養した場合,あるいはGFP-LC3を発現するトランスジェニックマウスを1日絶食させた場合,細胞内にGFP陽性の輝点を多数観察することができる104).しかしながら,GFP-LC3の輝点の数をオートファジー活性の指標とすることには問題もある.まず,LC3はオートファジーが阻害された条件下においてタンパク質の凝集体に取り込まれ,輝点として観察されてしまうので,これをオートファゴソームと誤認してしまう恐れがある105, 106)また,オートファゴソームとリソソームの融合を阻害するなど,オートファジーの後期ステップを阻害した場合にはオートファジーが抑制されているにもかかわらずオートファゴソームは増加してしまう62, 63, 65).そこで,オートファゴソームの形成からタンパク質の分解までのオートファジーの一連の流れ,すなわち「フラックス」を検出・定量することが重要になってくる53)(図4).
2)いろいろなオートファジーフラックスアッセイ
フラックスを評価する最も簡便な手法の一つは,リソソーム阻害剤を用いてLC3の分解を評価するLC3フラックスアッセイである.PE化してオートファゴソーム膜に結合したLC3は,電気泳動ではPE化されていないLC3より低分子量側に検出され,LC3-IIと呼ばれる107).オートファゴソーム内膜に結合したLC3はオートファゴソームとリソソームの融合に伴って分解されるので,リソソーム阻害剤で処理した場合としていない場合のLC3-IIのタンパク質量をウエスタンブロットで比較することで,オートファジーによる分解量を評価できる(図4A).しかしながら,この手法にも細胞や組織をすりつぶさなくてはならない,阻害剤あり・なしの2条件を常に用意しなければならない,ダイナミックレンジがあまり高くない,などの難点がある.また,リソソーム阻害剤の使用自体がオートファジーやLC3のPE化を誘導してしまうという知見もある53).ウエスタンブロットを用いたフラックスの評価には,オートファジーの選択的基質であるp62を検出・定量する手法もある(図4B).ただしこの場合,p62の発現量が転写レベルでの制御によって変動することにも注意する必要がある108).
リソソーム阻害剤を用いることなくオートファジー活性を用いる手法に,RFP(またはmCherry)とGFPとLC3をタンデムに接続した蛍光レポーターRFP-GFP-LC3[またはtandem fluorescent LC3(tfLC3)]を用いる方法がある109).GFPはリソソーム内の酸性環境で消光するが,RFPは消光しにくいため,オートファゴソームが黄色,オートリソソームが赤色の輝点として検出できる(図4C).この手法はLC3の代わりに特定のオルガネラに局在するタンパク質を用いることで,選択的オートファジーの検出にも応用することができる110–113).
筆者らはオートファジーフラックスを細胞および個体組織で簡便に評価するための蛍光レポーターとしてGFP-LC3-RFP-LC3ΔGを開発した114)(図4D).この蛍光レポーターは細胞内で発現するとATG4による切断を受け,GFP-LC3とRFP-LC3ΔGを等分子数生じる.GFP-LC3はC末端のグリシン残基(G)を介してPE化されるため,オートファジーによって選択的に分解されるが,RFP-LC3ΔGはほとんどが分解されずに細胞質に残留する.したがって,GFPとRFPの蛍光比を定量すれば,その細胞でオートファジーがどれだけ生じているかを評価することができる.RFP-LC3ΔGの部分はRFP単独でも同様に使用できる114).GFP-LC3-RFP-LC3ΔGを発現するトランスジェニックマウスの組織切片では,骨格筋においてオートファジーフラックスが活発な細胞とそうでない細胞を弱拡大の顕微鏡画像から判別することができた114).その他の有力なオートファジーフラックスアッセイとして,GFP-LC3の蛍光をフローサイトメトリーで評価する方法がある115)(図4E).この手法はGFP-LC3-RFP-LC3ΔGでも同様に適用でき,この場合RFPを発現量の内部標準として用いることができる.さらに筆者らは,培養細胞においてオートファジーをより簡便に評価する手法として,プレートリーダーを用いる手法を開発した.この手法では,96ウェルプレートにGFP-LC3-RFP-LC3ΔG発現細胞を播種し,それぞれのウェルを試薬添加などで処理した後,それを蛍光マイクロプレートリーダーにかけることでオートファジーフラックスを分析する114).これはオートファジー活性をハイスループットに評価できるという利点があり,筆者はこの手法でアミノ酸や血清の濃度の違いや,過去に報告されていたオートファジー誘導剤や阻害剤がオートファジーに及ぼす影響を網羅的に分析した114).ただし,このプレートリーダー法は相当感度が高い機材を使わないと蛍光を検出するのが難しい.2015年当時の時点で筆者が試した製品の範囲では,EnSpire, EnVision, SpectraMaxの3製品のみが明瞭にRFPのシグナルを検出できた.近年は画像取得も含めた「ハイコンテントスクリーニング」ができる顕微鏡も出ているので,それらの製品を用いるのも手だと思われる.なお,GFP-LC3の蛍光量でオートファジーを評価するこれらの手法は,オートファジーの誘導または阻害を開始してからGFP-LC3の十分な分解あるいは蓄積が生じてからでないとそれを検出できないことから,時間解像度が高くないという点には留意する必要がある.
オートファジーフラックスを評価する方法としては,このほかにKeimaを用いる方法がある116)(図4F).Keimaは中性条件下と酸性条件下で異なる励起ピークを持つ蛍光タンパク質である.このため,オートファジーにより細胞質からリソソームへの移動を,励起波長の違いとして検出することができる.RFP-GFP-LC3と同様,Keimaも特定のオルガネラに局在するタンパク質に付加して発現させることで選択的オートファジーの検出に用いることができる117, 118).
さらに最近,Halo-LC3またはHalo-GFPを用いるフラックスアッセイが開発された119, 120)(図4G, 4H).Haloタグは自己標識化タグの一つであり,リガンドとの間に共有結合を形成する.リガンドを結合させたHaloタグはリソソームにおける分解に強い抵抗性を持つことから,Halo-LC3がリソソームに運ばれるとLC3のみが分解され,ウエスタンブロットやゲル内蛍光イメージングでHaloタグが単体の分子量として検出できるようになる119)(図4G).HaloタグをLC3でなく特定のオルガネラに局在するタンパク質に付加しておけば,オルガネラの選択的オートファジーも検出することができる.さらに,Halo-GFPとして発現させた場合,タンパク質の非選択的な分解を検出することが可能である(図4H).Halo-GFPはATG結合系を欠損する細胞などにおいても,オートファジーによる分解を検出することができる119).
5. 個体におけるオートファジーの意義
1)オートファジーの役割の概要
オートファジー関連遺伝子を欠損するマウスの解析などにより,オートファジーはさまざまな役割を担っていることが明らかになってきている1, 10, 67, 102, 121–125).オートファジーの機能は大きく分けて細胞内成分の分解,および分解物の生成の二つに分けて考えることができる.
オートファジーによる分解は細胞内の品質管理に重要であり,オートファジーの抑制はポリユビキチン化タンパク質やp62を含む凝集体の蓄積,異常なミトコンドリアの蓄積などを通じて神経変性や肝障害を引き起こす126–128).また,肝臓でオートファジーを抑制したマウスでは多数の腫瘍形成がみられることから,オートファジーは腫瘍の抑制にも寄与していると考えられる129).また,オートファジーによるミトコンドリアの選択的分解は発生や分化の段階で重要な役割を果たしており,マイトファジーの抑制は赤血球の成熟を妨げることが報告されている130–132).
栄養飢餓によるオートファジーの誘導は,タンパク質の大規模な分解によりアミノ酸を産生することで恒常性維持に寄与していると考えられる.哺乳類においては,胎盤からの栄養供給が途絶える新生児期の飢餓において,オートファジーの活性化によるアミノ酸産生が重要な役割を果たしている133, 134).また成体においては,オートファジーは絶食時のアミノ酸の産生,血糖値の維持および生存に重要であることが報告されている135, 136).オートファジーは着床前の受精卵でも活性化されるが,このときのオートファジーはタンパク質の翻訳および受精卵の生存に重要である137).その他,オートファジーは脂肪滴の分解による脂肪酸の供給やフェリチンの分解による鉄の供給なども担っていることが報告されている121).オートファジーの生理的意義については,このほかにも多くの知見が得られてきているので,より詳しく知りたい方は別途各方面の総説を参照されたい1, 10, 67, 102, 121–125).
2)Atg遺伝子欠損マウスの表現型の違い
オートファジーの生理的意義の研究は,主にオートファジー関連遺伝子を全身または特定の臓器・組織において欠損するマウスによって行われてきた.初期にはAtg5やAtg7のノックアウトマウス(以下KOマウス)が主に用いられていたが,近年ではほかのKOマウスを用いた研究も多く出てきている.しかしながら,さまざまなマウスが作製されるにつれて,異なるAtg遺伝子の欠損が異なる表現型を呈する事例もみられるようになってきている.
表1に,これまでに報告されている全身KOマウスとその表現型をまとめた.これらのKOマウスは死亡する時期によって大きく4グループに分類することができる.早期に死亡するものから順に,第1のグループは胎生8.5日目までに致死となるPik3r4(Vps15),Becn1(Beclin 1),およびPik3c3(Vps34)のKOマウス138–140),第2のグループはおおむね胎生13.5日目から18.5日目までの間に致死となるAtg9a, Rb1cc1(Fip200),Atg13,およびAtg101 KOマウス15, 141–143),第3のグループは出生後1日以内に致死となるAtg3, Atg5, Atg7, Atg12, Atg16l1のKOマウスおよびUlk1/Ulk2, Wdr45b/Wdr45(Wipi3/Wipi4)のダブルKOマウス126, 133, 144–149),第4のグループは成体まで生き残るAtg4b, Atg4c, Atg4d, Map1lc3b(Lc3b),Gabarap, Ulk1, Ulk2, Wdr45b, Wdr45, Atg2b, Nrbf2のKOマウス,およびAtg4c/Atg4dのダブルKOマウスである18, 131, 149–156).なお,Atg14のKOマウスは胎生致死であったとは述べられているものの,何日目までに死亡するか示されていないため,グループ1と2のどちらに含まれるのか不明である156).これら一連のマウスの表現型の違いを理解するには「オートファジーにおける必要性」および「オートファジータンパク質が持つ非オートファジー機能」の二つの観点が必要になる.まず成体まで生存する第4のグループについてだが,これは当該遺伝子のオートファジーにおける必要性が低かったために,KOマウスにおいてオートファジーがおおむね正常に起こり,グループ3の新生児期の致死が生じなかったものと考えられる.具体的には,ある遺伝子の機能がそのパラログによって補完されるケースが考えられる.たとえばAtg2についてはAtg2a, Atg2bどちらかを欠損した細胞ではオートファジーには影響がみられず,両方を欠損した際に初めてオートファジーが強く抑制される157).ULKについても同様で,Ulk1のKOでも多少の影響はあるものの,Ulk1, Ulk2両方をKOした場合にオートファジーの抑制が強くなる17).Atg4, Wdr45, lc3b, Gabarapについても,パラログによる機能の補完が表現型を出にくくしているものと推察される.ただし,組織によってはパラログによる補完が十分に機能せず,オートファジー活性が低下するケースもあるようだ.たとえばUlk1 KOマウスでみられる赤血球の成熟不全は,造血細胞特異的にAtg7を欠損するマウスの表現型が弱く出ているものと考えられるし131, 132),Wdr45b KOマウスの神経変性の表現型は神経特異的にAtg5やAtg7を欠損したマウスの表現型に類似している127, 128, 149).
表1 オートファジー関連遺伝子ノックアウトマウスとその表現型| 分類 | 遺伝子名 | 生存期間(死亡時期) | 観察されたその他の表現型 | 文献 |
|---|
| グループ1 | Pik3r4 (Vps15) | E7.5以前 | — | 138 |
| Becn1 (Beclin 1) | E8.5 | 成長遅延,胚葉形成不全,細胞死 | 139 |
| Pik3c3 (Vps34) | E8.5 | 成長遅延,胚葉形成不全 | 140 |
| グループ1 or 2 | Atg14 | 詳細不明(胎生期) | — | 156 |
| グループ2 | Atg9a | E13.5–E17.5(一部新生仔期) | 成長遅延 | 141 |
| Rb1cc1 (Fip200) | E14.5–E16.5 | 心筋層の菲薄化,肝臓の異常 | 142 |
| Atg13 | E17.5–E18.5 | 成長遅延,心筋層の菲薄化 | 15 |
| Atg101 | E17.5–E18.5 | 成長遅延 | 143 |
| グループ3 | Ulk1/Ulk2 | E18.5–新生仔期(生後1日以内) | 脳梁と前交連の軸索の低形成 | 148 |
| 新生仔期(生後1日以内) | 肺胞隔壁の肥厚,肺胞上皮細胞でのグリコーゲン蓄積 | 147 |
| Wdr45b/Wdr45 (Wipi3/Wipi4) | 新生仔期(生後1日以内) | 成長遅延,神経細胞での凝集体蓄積 | 149 |
| Atg3 | 新生仔期(生後1日以内) | 形態学的異常なし,成長遅延ありアミノ酸濃度の低下,吸啜障害 | 144 |
| Atg5 | 新生仔期(生後1日以内) | 133 |
| Atg7 | 新生仔期(生後1日以内) | 126 |
| Atg12 | 新生仔期(生後1日以内) | — | 146 |
| Atg16l1 | 新生仔期(生後1日以内) | 成長遅延 | 145 |
| グループ4 | Atg4b | 成体まで生存 | 運動失調,小脳深部核・前庭核でスフェロイド様構造形成 | 140 |
| Atg4c | 成体まで生存 | 絶食時活動量低下,横隔膜筋線維の構成変化,リンパ球減少,発がん物質による腫瘍形成増加,脾臓での細胞死 | 151 |
| Atg4d | 成体まで生存 | プルキンエ細胞の脱落,運動能力低下,リンパ球減少 | 151 |
| Atg4c/Atg4d | 成体まで生存 | プルキンエ細胞の脱落,運動能力低下,リンパ球減少,心拡張 | 151 |
| Map1lc3b (Lc3b) | 成体まで生存 | 目立った異常なし | 152 |
| Gabarap | 成体まで生存 | 目立った異常なし | 153 |
| Ulk1 | 成体まで生存 | 網状赤血球数の増加,ミトコンドリア除去の遅延 | 131 |
| Ulk2 | 成体まで生存 | 目立った異常なし | 18 |
| Wdr45b (Wipi3) | 成体まで生存 | 成長遅延,運動失調,認知機能障害,長期増強の減弱,神経軸索スフェロイド,プルキンエ細胞の脱落 | 149 |
| Wdr45 (Wipi4) | 成体まで生存 | 認知機能障害,てんかん感受性,海馬CA1神経細胞のシナプス後応答の低下,前頭皮質および黒質における神経細胞の脱落 | 154 |
| Atg2b | 成体まで生存 | 目立った異常なし | 155 |
| Nrbf2 | 成体まで生存 | 肝臓での細胞死 | 156 |
| これまでに報告されたオートファジー関連遺伝子のノックアウトマウスとその表現型のまとめ.全身で遺伝子を欠損するマウスのみ記載し,コンディショナルノックアウトマウスについては含めていない. |
では,より厳しい表現型を示すグループ1/2のマウスはどう解釈できるだろうか? これは「オートファジータンパク質が持つ非オートファジー機能」によって解釈できる可能性がある158).たとえばATG14以外のクラスIII PI3K複合体構成因子はエンドサイトーシスにおいても機能しているので,そちらの機能の欠損がグループ1のマウスの早期の胎生致死をもたらした,と推測することは可能であろう158).またFIP200についてはさまざまなタンパク質と相互作用し,複数のシグナル伝達経路に影響を与えることが報告されているため142, 159–161),Rb1cc1のKOがそれらのシグナルの異常をもたらすことで胎生致死を招いており,ATG13, ATG101を含めた,ただしULK1/2を除いた複合体がFIP200の「非オートファジー機能」に関与している,という推測も一応は可能である.しかし,さまざまなマウスの情報が得られてきた現時点では,筆者は別の可能性をより強く考えている.グループ3のマウスの表現型がグループ2のマウスの表現型より弱いのは,オートファジーが部分的に残っていたから,という可能性である.たとえばATG結合系の遺伝子をKOした細胞ではオートファジーが生じうることが示されており55),Halo-GFPによってフラックスも検出されている119).Ulk1/Ulk2, Wdr45b/Wdr45のダブルKO細胞についても,同様にオートファジーフラックスが弱いレベルで検出されている17, 33).このわずかなオートファジーが胎生致死を回避させた,という可能性は十分に考えられる.ATG13結合部位を変異させたFIP200を発現するノックインマウスが胎生致死を回避して新生児期に致死となるという報告もあるが162),これもATG13との結合が部分的に残っているなどしてオートファジー活性がわずかに回復したから,と考えれば矛盾なく解釈しうる.
筆者はAtg13およびAtg101のノックアウトマウスを作製・解析し,これらが胎生致死となることを示したが15, 143),あるいはこれこそが「オートファジーを完全に止めたことによる表現型」だったのかもしれない.なお,一般に胎生12.5日以降のマウスが致死となる主要な要因は胎盤,血管,心臓,または血液の異常であるが163),Atg13 KOマウスでは胎生15.5日目から17.5日目にかけて心筋層の菲薄化(ひはくか)が認められた15).一方で胎盤,血管,血液に関しては明確な組織学的異常が認められなかったことから,Atg13 KOマウスは心不全により死亡しているものと考えられる.この心筋層の菲薄化はFip200 KOマウスでも観察されている142).もしオートファジーが胎児の心臓の発達に重要であるとするならば,そのメカニズムは何なのか,興味深いところである.
6. オートファジーが関連する疾患とオートファジー制御薬
1)オートファジーが関連する疾患と治療戦略
オートファジーは多くの疾患に関与していることが明らかになっている2, 67, 102, 122, 123, 125, 164, 165).詳細はほかの文献に譲ることとして,ここでは神経変性疾患とがんの2点に焦点を当てる.アルツハイマー病,パーキンソン病,ハンチントン病などの神経変性疾患では,共通する特徴として細胞内タンパク質のミスフォールディングと凝集が観察され,多くの場合こうしたタンパク質の蓄積が病態に関係していると考えられている165, 166).動物実験では,神経特異的オートファジー欠損マウスがタンパク質凝集体の蓄積や神経細胞の脱落,運動失調などを示しており,オートファジーと神経変性疾患の関連が示唆されてきた127, 128)が,近年,患者から得られた情報を通じて,オートファジーと神経変性疾患の関係がさらに明らかになりつつある.たとえば,SENDA(static encephalopathy of childhood with neurodegeneration in adulthood)の患者においては,WIPI4をコードする遺伝子WDR45にオートファジー活性を低下させる変異が見つかっている33, 167).また小児型運動失調症の患者でも,オートファジー活性の低下を伴う変異が報告されている168).さらに90名のアルツハイマー病患者の死後脳の前頭皮質のプロテオーム解析では,患者の脳でp62の蓄積が認められており,オートファジー活性の低下が示唆されている169).そしてパーキンソン病にはPINK1-Parkin経路を介したマイトファジーの破綻が関与していると考えられる67, 125, 165).これらのことから,神経変性疾患においてはオートファジーの活性化が治療戦略として考えられている164).がんとオートファジーの関係については,オートファジーが細胞の腫瘍化を抑制する一方で,腫瘍化した細胞の生存にはオートファジーがしばしば正に作用することがわかっている170).実際に,オートファジーの抑制が腫瘍細胞の生存を妨げることが示されている136).したがって,がん患者においては,腫瘍のオートファジーを抑制する治療戦略が検討されている164, 171).
2)臨床で使用可能なオートファジー制御薬の探索
現時点では,mTORC1の阻害剤によるオートファジー誘導やリソソーム阻害剤によるオートファジー抑制が可能であり,これらを用いた疾患治療を目的とした臨床試験も行われている172, 173).しかし,これらはオートファジー以外にも影響を及ぼし,深刻な副作用をもたらす可能性がある.mTORC1阻害剤は免疫抑制作用を持ち,リソソーム阻害剤は毒性が高いことが報告されている172, 173).安全性の高いオートファジー制御薬の開発は依然として重要な課題である.これまでに,オートファジーを制御する薬剤・化合物のスクリーニングがさまざまな手法で試みられ,mTORC1やリソソームに依存しない薬剤も複数見つかっている174).安全な薬剤の探索においては,既存薬を新たな治療用途に活用する「ドラッグ・リポジショニング(またはドラッグ・リプロファイリング)」が有効な戦略である175).筆者らはGFP-LC3-RFP-LC3ΔG発現細胞を用いたハイスループット測定により,1054種類の既承認薬を網羅的に分析し,47個のオートファジー誘導剤と43個の阻害剤を同定した114).これらにはmTORC1やリソソームを抑制するものや毒性が高いと考えられる薬剤が多く,安全なオートファジー制御薬としての即時実用化は難しいと思われた.しかし,同様のアプローチでより安全性の高い薬剤を探索することは十分可能であると考えられる.なお,筆者らが同定した43個のオートファジー阻害剤には,過去にオートファジーの「誘導剤」として報告されていた薬剤が10個含まれていた.これはオートファジー後期ステップの阻害によるオートファゴソームの蓄積が「オートファジーの活性化」と誤認されていた可能性が考えられる.よってオートファジー制御薬の探索を行う際には,やはりGFP-LC3-RFP-LC3ΔGなどオートファジーのフラックスを評価できる手法を用いることが推奨される.
1. 神経科学の潮流
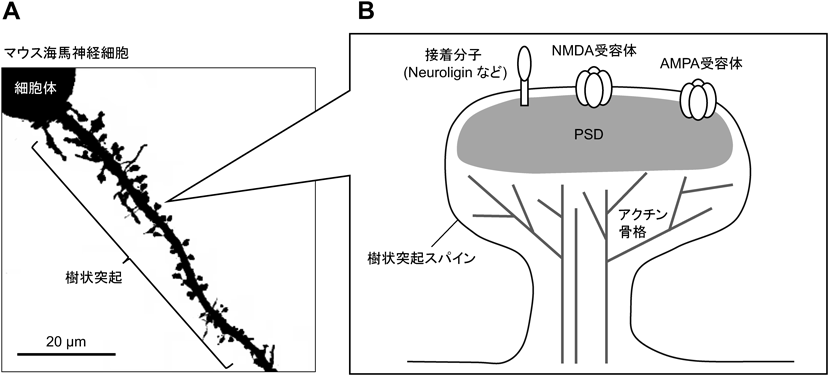
生化学がそうであるように,神経科学もまた広大な学問分野である.まず,ミクロからマクロまでのさまざまな観点が存在する.mRNAやタンパク質などの分子に焦点を当てた研究もあれば,神経細胞やグリア細胞の形態や機能の研究もある.そして神経細胞間の接続により形成される神経回路の研究もあれば,脳の組織ないし臓器全体を分析する研究もある.そして——ここからが神経科学の特徴的な要素になるが——個体や集団の認知や行動に主軸を置いた研究も,分野の中でかなりのウエイトを占めている.また,細胞から臓器のレベルにおいては,脳やその構成成分を物質としてとらえ,分析する「生化学・組織学」の視点と,神経細胞の電気的な活動パターンを単一細胞レベルから脳全体までさまざまな規模で分析する「生理学」の視点が存在する.近年のトレンドとしては,個体の行動や認知に関わる神経回路を特定し,その機能を光遺伝学などで分析する生理学的研究や,二光子顕微鏡・透明化などの技術を駆使したイメージング,個々の細胞の遺伝子発現パターンを網羅的に分析するトランスクリプトーム研究,そして神経細胞による脳の各部位の接続を網羅的に分析する「コネクトーム」などがあるようだ176–179).筆者は2016年に神経科学の研究に参画したが,多くの切り口がある中で,タンパク質に主軸を置いた研究は分野全体からみると比較的マイナーであり,まだまだ開拓の余地があるのではないかと思われた.それでは,どういったタンパク質を研究するか?と考えた結果,着目したのが「シナプス後肥厚(postsynaptic density:PSD)」を中心としたシナプスのタンパク質群である.ここではシナプスのタンパク質の研究について,筆者の最近の仕事を交えつつ解説する.
2. シナプス後肥厚(PSD)を構成するタンパク質群
1)PSDの主要なタンパク質
興奮性シナプスの多くは,神経細胞の樹状突起上に形成される突起上の構造体である樹状突起スパインの上に形成される180–182)(図5A).成熟した樹状突起スパインはきのこ型の構造を持っており,この形状はアクチン骨格によって支えられている.PSDは樹状突起スパイン先端部の細胞膜直下に形成されるタンパク質の集積構造であり,もともとは電子顕微鏡で観察される高密度の構造として観察されたことからこのように命名された183).PSDには1000種類以上のタンパク質が集積しており,シナプスの構造や機能において重要な役割を果たしている184–186)(図5B).また,自閉症や統合失調症で変異が見つかっている遺伝子の中には,PSDに局在するタンパク質をコードするものが多く含まれている187–189).NMDA(N-methyl-D-aspartic acid)受容体やAMPA(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)受容体などのイオンチャネル共役型グルタミン酸受容体は,シナプス前部から放出されるグルタミン酸の結合により陽イオンを流入させ,興奮性シナプス後電位を発生させる.PSDに豊富に含まれるPSD95(postsynaptic density protein 95)などの足場タンパク質は,グルタミン酸受容体を含むさまざまな後シナプスタンパク質と結合し,後シナプスのタンパク質相互作用ネットワークを形成している.Neuroligin, N-cadherinなどの細胞接着分子はシナプス前部の接着分子と相互作用することでシナプスを形成している.また,PSDにはCaMKII(Ca2+/calmodulin-dependent protein kinase),SynGAP(synaptic GTPase-activating protein)などのシグナル伝達酵素が局在しており,これらの多くはNMDA受容体を介したカルシウムイオンの流入によってその活性が調節される186).近年,これら主要なPSDタンパク質は液-液相分離によって後シナプスに集積することが示されている190–192).
2)プロテオーム解析によるPSDのタンパク質組成の解明
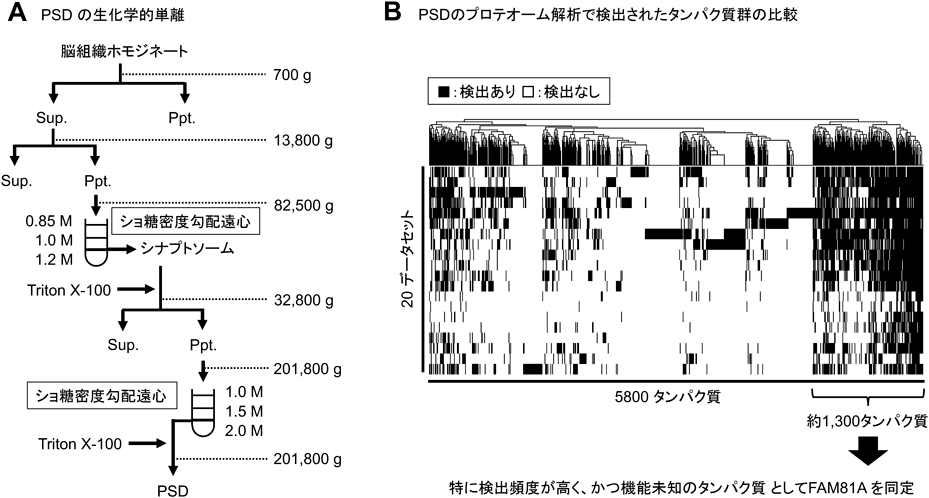
PSDは生化学的に単離することができ,その手法の基礎は1980年に確立している193).分画遠心とショ糖密度勾配遠心により,シナプスの膜画分である「シナプトソーム」を分離し,界面活性剤Triton X-100で処理した上で遠心すると,PSDが不溶性画分として得られる(図6A).この手法はその後多少の改良が施されてはいるものの,基本的には現在でもPSDを単離するために広く用いられている.こうして単離したPSDについて質量分析によるプロテオーム解析を行うと,PSDに局在するタンパク質を網羅的に同定することができる.初期の研究で検出されたタンパク質は数十個から数百個であったが,2011年と2012年に実施されたヒトおよびマウスの脳のPSDプロテオーム解析では,いずれにおいても1000種類強のタンパク質が検出されている194, 195).その後も質量分析の感度の向上に伴い,検出されるタンパク質数はさらに増えてきたが,そうなってくると,今度はサンプル調製時における微量の夾雑物など,PSD以外のタンパク質も検出されてしまうことが懸念される.筆者らは,これまでに報告されたPSDプロテオームのデータ群を統合することにより,共通して検出されているシナプスタンパク質の抽出を試みた192).PSD画分のプロテオーム解析では,20セットのデータで検出された計5800タンパク質のうち,約1300のタンパク質が複数のデータで共通して検出されていたことから,これらがPSDを構成するコンセンサスタンパク質群であると考えられる192)(図6B).なお,別の研究グループはプレシナプスも含めた,さらに包括的なシナプスのプロテオミクスのデータベースを作成している196).
3)主要PSDタンパク質FAM81Aの同定
PSDに1000種類以上のタンパク質が局在するのであれば,その中には重要であるがまだ研究されていないタンパク質も少なからず含まれていると考えられる.筆者らは最近,20セットのPSD画分のプロテオームおよび15セットのPSDタンパク質結合因子のプロテオームのデータを統合し,繰り返し検出されているにもかかわらず機能が未知であったタンパク質を洗い出した.その結果,最もヒット数の多かったタンパク質としてFAM81A(family with sequence similarity 81 member A)が同定された192).このタンパク質は計21のデータセットで検出されており,免疫電子顕微鏡法でもPSDに局在することが報告されていた197)ものの,その機能・性質についてはほとんど何もわかっていなかった.
培養神経細胞などを用いた解析の結果,FAM81Aが自己会合し,液-液相分離によって液滴を形成することでPSDへと局在すること,そしてPSD95, SynGAP,およびNMDA受容体サブユニットのGluN2Bという三つの主要なPSDタンパク質と結合することがわかった192).また,FAM81Aはin vitroでこれらの主要PSDタンパク質とともに液滴を形成し,その液滴を拡張させる作用を持っていた.FAM81Aを過剰発現すると樹状突起スパインが拡張し,ノックダウンするとPSDが縮小したことから,FAM81Aは主要なPSDタンパク質を集積させる,あるいはそれらの散逸を防ぐことでシナプスのサイズを正に制御する因子であると考えられる.
FAM81Aがほかの主要PSDタンパク質と比べて独特なのは,その進化的な「新しさ」である.哺乳類FAM81Aには精巣でのみ発現するFAM81Bというパラログがあるが,FAM81A/Bの両方を持っているのは脊椎動物では哺乳類,鳥類,爬虫類のみであり,両生類,魚類はFAM81Bのみを持っている192).そして,魚類のPSDではFAM81Bは検出されていない198).よって,FAM81Aは高等動物のシナプスに特徴的なタンパク質であるといえるだろう.
3. プロテオミクスからとらえるシナプスの成熟
1)生後発達期のシナプスの成熟
生後発達期におけるシナプスの成熟は脳の発達において重要であり,このプロセスの異常は自閉症や統合失調症と関係している181, 199).シナプスの数は,初期には増加していき,その後やや減少する186, 199).発達後期におけるシナプスの減少は,シナプスの刈り込みと呼ばれている.ただし,これは単に増加して減少するというよりも,後述のように継続的に生じている形成と除去のバランスが徐々に変化していく結果であると考えられる.樹状突起スパインの形状は,初期には細長い形状であるのが,徐々に頭部が拡張してきのこ型へと成熟する180).また,幼少期は樹状突起スパインの形成と除去が頻繁に生じているのが,成長とともに徐々に安定化していく180, 186).発達期のシナプスでは,タンパク質レベルでも何らかの変化が生じていると考えられる.最近,筆者らおよびKriegsteinらのグループは,生化学的に単離したPSDの定量的プロテオーム解析により,生後発達期におけるPSDのタンパク質組成の変化を明らかにした200, 201).発達期のマウスのPSDで顕著な増減を示すタンパク質の中には,発現量に応じてシナプスの数や形状が変化する因子が多く含まれていたことから,PSDのタンパク質組成の変化はシナプスの成熟に関与していると考えられる.パスウェイ解析ではアクチン骨格の再編成を制御するRho GTPaseシグナルの関連のタンパク質が濃縮されていたが,実際,シナプトソームにおいては発達に伴ってRho GTPaseシグナルが抑制されていることがわかった201).これらの結果から,発達期のPSDのタンパク質組成の変化は後シナプスのシグナル伝達の変化などを介してシナプスの成熟に寄与しているのではないかと考えられる.
2)げっ歯類と霊長類のシナプス発達の違い
発達後期におけるシナプスの刈り込みは広く知られている現象であるが,実際に筆者が個々の論文のデータにあたってみると,実はマウスやラットの大脳皮質では,発達期に増加したシナプスの数はその後ほとんど減少していないようだった186).これに対し,ヒト,マカク,チンパンジー,コモンマーモセットなど霊長類の大脳皮質では,実に半分近くのシナプスが刈り込まれているようだった186).すなわち,げっ歯類と霊長類ではシナプス発達のパターンが異なっていると考えられる.そこで筆者らは,生後発達期のコモンマーモセットのPSDのプロテオーム解析を行い,マウスとの違いを分析した201).その結果,2週齢以降のマウスの脳で生じる変化は,コモンマーモセットではおおむね出生直後から生後2か月までの早い段階で生じており,それ以降はさらに別のタンパク質の増減が生じていることがわかった.この発達後期に生じるタンパク質群の中にも,シナプス形成シグナルに関連するタンパク質群がエンリッチしていたことから,2か月齢以降に生じるPSD組成の変化が,コモンマーモセットの脳で3か月齢以降に生じる202)顕著なシナプスの刈り込みに関与している可能性は十分に考えられるといえるだろう.
4. プロテオーム解析からシナプトーム解析へ
1)個々の,そして脳全体のシナプスの分析
プロテオーム解析は異なる脳の部位や年齢,生物種におけるシナプスのタンパク質組成の違い,生理的条件におけるシナプスの変化などの研究に広く用いられてきた185, 186).しかしこの手法には根本的な題がある.脳をすりつぶしてPSDなどをバルクで抽出してくる過程で,失われてしまう情報が出てくるのである.たとえばシナプスのサイズはシナプス伝達の強度に関係する重要な指標であるが182),この情報はプロテオーム解析では読み取れない.さらに,個々のシナプスのタンパク質組成の違いを評価できないという問題もある.
Seth Grant博士の研究室は長年シナプスのプロテオーム解析を展開してきたが,近年,イメージングにより個々のシナプスを全脳スケールで分析する「シナプトーム」というアプローチを提唱している203).この方法では,まず主要シナプスタンパク質に蛍光タンパク質を付加したノックインマウスから脳切片を作製し,シングルシナプスレベルの解像度で全体をスキャニングする.通常は1切片あたり数時間から十数時間をかけて2万枚以上の画像を取得する形になる.次に,機械学習を用いた画像処理により個々のシナプスを抽出して分析する.PSDの主要足場タンパク質PSD95およびSAP102(synapse-associated protein 102)に蛍光タンパク質を付加したノックインマウスのシナプトーム解析では,脳の部位や週齢によるシナプスの大きさ,形状,タンパク質組成の違いが明らかになっている203, 204).さらに同研究グループは,この手法により特定の遺伝子の欠損や断眠が,脳のどの部位のシナプスにどのような影響を及ぼすかも明らかにしている203, 205, 206).シナプトーム解析は一度に限られた数のタンパク質しか分析できない代わりに,プロテオーム解析では失われてしまう多くの情報を得られる手法であるといえるだろう.一連の研究で興味深い点の一つは,PSD95単独陽性,SAP102単独陽性,そして両タンパク質陽性のシナプスが脳の各部位で観察されていることである.一般に,脳にはAMPA受容体を欠く「サイレントシナプス」があることが知られており207),筆者の研究ではFAM81A陽性のシナプスと陰性のシナプスが観察されている192).これらの知見をまとめると,シナプスのタンパク質組成は実は個々に相当異なっているのではないかと考えられる.総説などではしばしば,主要なシナプスタンパク質群が一つのシナプス内に共存しているような図が描かれているが,この概念は今後修正する必要があるかもしれない.シナプトーム解析はシナプスのタンパク質組成の,ひいては性質や機能の多様性を明らかにしていく上でも重要なアプローチであると考えられる.
2)シナプスの微細構造の網羅的解析
シナプスの微細構造,すなわちナノメートルスケールでのタンパク質配置は,近年のシナプス研究のホットトピックの一つである.シナプスにおいてタンパク質がランダムに分布しているわけではないことは電子顕微鏡を用いた研究からも示唆されていたが208),近年は超解像顕微鏡によるシナプスのタンパク質分布の研究が発展してきている209).PSDにおいて,PSD95はAMPA受容体とともにナノクラスターと呼ばれる部分的にタンパク質が密集した微細構造を形成している210).このPSD95-AMPA受容体のナノクラスターはRIM1/2(Rab3-interacting molecule 1/2)が局在するシナプス小胞の放出部位の正面に位置することでシナプス前後にわたる「ナノカラム」構造を形成している211).ナノカラムにおけるシナプス前後の構造は接着分子であるNeuroligin 1やLRRTM2(leucine rich repeat transmembrane neuronal 2)によって接続されているが,これらの接着分子に介入してナノカラム構造を崩すとシナプス伝達の効率が低下することから,ナノカラムなどのシナプスの微細構造は,機能的に重要な意味を持っていると考えられる212, 213).これらの研究は主に培養神経細胞を用いて行われているが,マウス個体の脳でもPSD95のナノクラスターは観察されており,脳の部位によってナノクラスターの数や構造に違いがあることが示唆されている214, 215).筆者らはシナプスの微細構造についてシナプトーム解析を行おうと考えたが,超解像顕微鏡はスループットやデータサイズなどの面で,全脳のシナプスの分析に適用するのは難しい.そこで筆者らは,PSD95タンパク質どうしの近接度をFRET(Förster resonance energy transfer)で評価し,これを指標としてシナプトーム解析を行った.その結果,脳の部位や年齢により,シナプスの微細構造に違いがあることが示唆された(筆者ら 投稿準備中).詳細は近日中に論文として発表する予定なので,興味を持たれた方はぜひご覧いただければ幸いである.