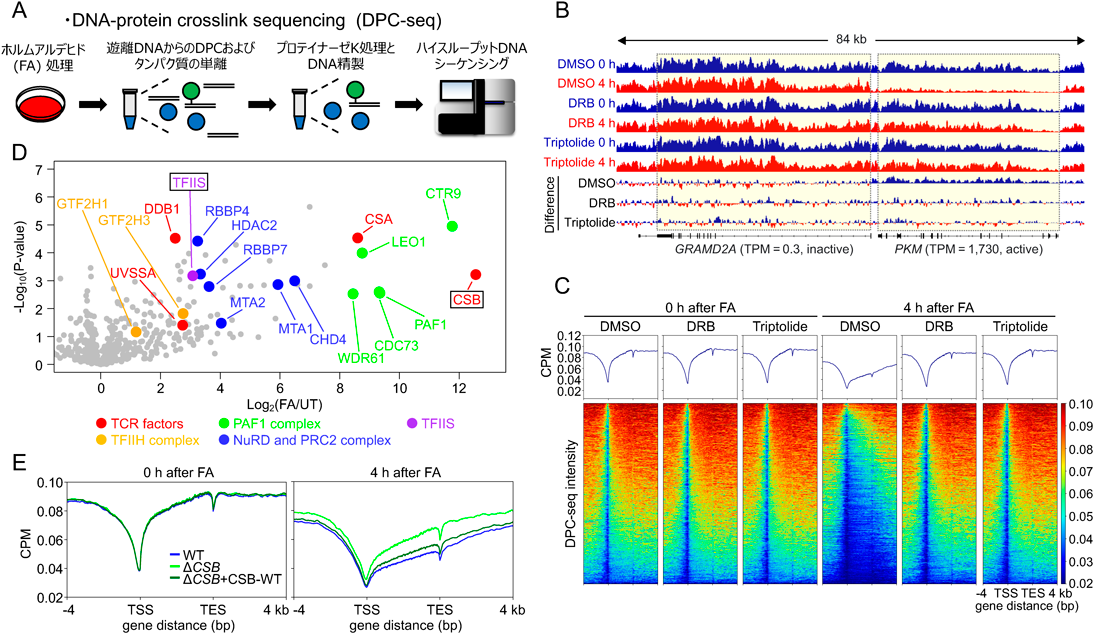
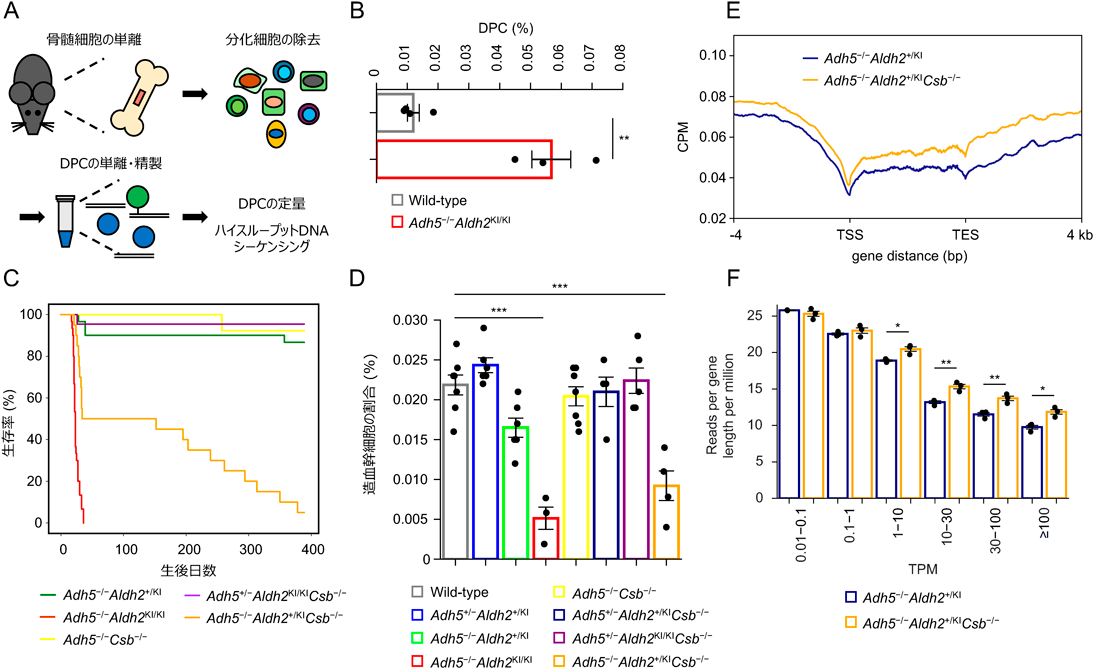
内在性のアルデヒドに由来するDNA損傷の修復機構とその破綻により発症する遺伝性疾患についてRepair mechanisms of endogenous aldehyde-induced DNA damage and associated human disorders
1 東海国立大学機構 名古屋大学環境医学研究所発生・遺伝分野Department of Genetics, Research Institute of Environmental Medicine (RIeM), Nagoya University ◇ 〒464–8601 愛知県名古屋千種区不老町 ◇ Furo-cho, Chikusa-ku, Nagoya, Aichi 464–8601, Japan
2 東海国立大学機構 名古屋大学医学系研究科人類遺伝・分子遺伝学教室Department of Human Genetics and Molecular Biology, Nagoya University Graduate School of Medicine ◇ 〒464–8601 愛知県名古屋千種区不老町 ◇ Furo-cho, Chikusa-ku, Nagoya, Aichi 464–8601, Japan
3 東海国立大学機構 One Medicine創薬シーズ開発・育成研究教育拠点動物医科学研究開発部門Division of Animal Medical Science, Center for One Medicine Innovative Translational Research (COMIT), Nagoya University ◇ 〒464–8601 愛知県名古屋千種区不老町 ◇ Furo-cho, Chikusa-ku, Nagoya, Aichi 464–8601, Japan
4 東海国立大学機構 糖鎖生命コア研究拠点分子生理・動態部門Division of Molecular Physiology and Dynamics, Institute for Glyco-core Research (iGCORE), Nagoya University ◇ 〒464–8601 愛知県名古屋千種区不老町 ◇ Furo-cho, Chikusa-ku, Nagoya, Aichi 464–8601, Japan