日本および諸外国における脳血管疾患の頻度は高く,国内の総患者数は174.2万人1),死亡原因の第4位である2).また米国での有病率は3.4%である3).脳血管疾患は大別すると脳梗塞などの虚血性脳血管障害と脳内出血,くも膜下出血などの出血性脳血管障害があり,その半分以上を脳梗塞が占める.後遺症によるQOL低下が著しく,少しでも後遺症の軽減につながる治療法の確立は重要な課題である.
現在の脳梗塞治療の柱は,①超急性期の血栓溶解療法や血管内治療といった再灌流療法,②急性期の酸化ストレスや脳浮腫に対する脳保護療法,③急性期~慢性期の抗血小板剤による再発予防,④リハビリテーションである.超急性期治療の標的は,梗塞周辺部の,虚血により機能障害に陥っているが細胞死には至っていない神経細胞が存在する低灌流領域(ペナンブラ領域)である.脳梗塞が完成する前に再灌流療法を行うことで病態の改善が期待できるものの,ほとんど障害を残さず回復するのは約1/3である4).さらに,治療適応を判断するための検査に時間がかかることや,早期再閉塞や出血合併症など治療特有の問題もあり,これらの改善が求められている.血栓溶解療法に先立つ抗酸化剤投与の効果を示す基礎研究やコホート研究もあるが,臨床上の有効性は証明されていない.
Gタンパク質共役型受容体(G protein-coupled receptor:GPCR)は,細胞膜あるいは細胞小器官の構成膜上に存在する受容体の一種である.膜を7回貫通すること,Gタンパク質を介さないシグナル伝達も担うことから,7回膜貫通型受容体と呼ぶことも提唱されている.通常,GPCRのN末端は細胞外に,C末端領域は細胞内に位置する.細胞外の神経伝達物質やホルモン,アミノ酸や,光子といった物質(リガンド)を受容すると,受容体は構造変化を起こし,細胞内において2種類のシグナル経路(三量体Gタンパク質あるいはβアレスチンを介したシグナル経路)が活性化される.三量体Gタンパク質を介したシグナル経路では,細胞内側に結合している三量体Gタンパク質のαサブユニットが,GDP結合型からGTP結合型へと変換される.変換されたGタンパク質は,次にエフェクターの活性を変化させて,細胞外の変化を細胞内へと伝達する.βアレスチンを介したシグナル経路では,リガンドによりGPCRが活性化されると,GRKなどのキナーゼによってリン酸化されてβアレスチンが結合する.このβアレスチンの結合により,GPCRが脱感作されるとともに,β-アレスチン依存的なシグナル伝達が活性化される.米国食品医薬品局(FDA)承認薬のおよそ30%以上がGPCRを標的としていることや,わが国の医薬産業政策研究所の調査において開発薬全体の20%(253/1265)がGPCRを標的としていることから,現在においても依然としてGPCRが有力な創薬ターゲットであることがわかる.
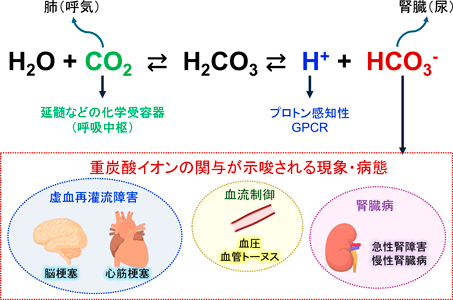
最近筆者らは,血管壁細胞(ペリサイトや血管平滑筋細胞)に発現するGPCRであるGPR30が重炭酸イオンによってpH非依存的に活性化されることを見いだし,さらにGPR30が血流を制御して虚血再灌流障害に関与することを明らかにした.次節では,その内容を紹介する.
3. 重炭酸イオンによるGタンパク質受容体活性化と虚血再灌流障害への関与
1)研究の背景(図1)
生体内のpHは,炭酸・重炭酸緩衝系(H2O+CO2⇄H2CO3⇄[H+]+[HCO3−])をはじめとする複数の緩衝系によって,7.4付近に保たれている.しかし,分泌細胞周囲や腫瘍内部,虚血・再灌流部位など,局所的にはpHやイオン濃度の大きな変化が存在する.これまで,プロトン(H+)を感知するGPCRが複数発見され5, 6),pHの低下がGPCRを介して細胞応答を引き起こすことが知られていた.一方,近年,重炭酸イオン(HCO3−)や二酸化炭素も細胞応答を引き起こすことを示唆する報告があり7, 8),これらを認識する受容体の存在が示唆されていた.しかしながら直接的な証明はされておらず,そのメカニズムも不明であった.
虚血再灌流障害は脳梗塞や心筋梗塞などでみられ,微小循環障害を主体とする病態である9).急激な血行動態変化によるpHやイオン濃度の変化,酸化ストレス等の強いストレスに細胞がさらされる結果,さまざまな細胞応答が生じる.虚血再灌流障害には脳微小血管壁細胞の収縮の関与が示唆されているものの,その機序は未解明である.重炭酸イオンがpH変化とは異なる機序で虚血再灌流障害を悪化させるという報告もあり10),酸塩基平衡の変化に応答する新規のシグナル伝達経路の存在が示唆されていた.そこで筆者らは,酸塩基平衡の変化に応答する新規のシグナル伝達経路があるのではないかと考え,酸塩基平衡に関与するGPCR候補を探した.
2)GPR30は重炭酸イオンによって活性化する
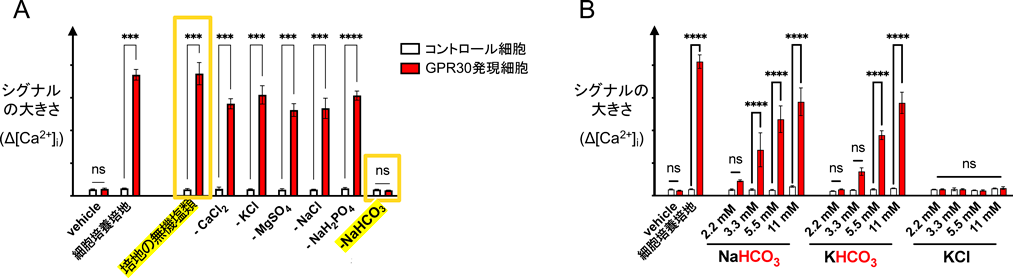
まず,GPCRの組織発現量および脳微小血管の一細胞RNAシーケンスのパブリックデータベースを用い,酸塩基平衡に関連し,かつ脳微小血管に発現するGPCR候補としてGPR30に着目した.GPR30はエストロゲンによるnon-genomic actionを担うと報告された膜型エストロゲン受容体である11).エストロゲンの作用としては,核内受容体ERを介した転写促進作用がよく知られている.一方,エストロゲンが転写を介さないすばやい応答を引き起こすこと,それを担うのがGPR30であることが2000年代前半に報告された.そこでエストロゲンによるGPR30活性化を検証しようとしたが,筆者の実験系ではエストラジオールやタモキシフェンといった既報のリガンドへの反応は認められなかった.そこで筆者は,GPR30の新規リガンドを探索することを考え,その過程で,GPR30安定発現細胞では細胞培養培地(DMEM)の添加によって細胞内カルシウムが上昇することを偶然発見した.そして培地中の各成分を添加してGPR30の応答を解析することで,GPR30が培地中の重炭酸イオンに応答していることを突き止めた(図2)12).さらに,GPR30は生理的濃度(EC50=11~12 mM)の重炭酸イオンによって活性化されること,下流シグナルとしてイノシトールリン脂質代謝の亢進やERKのリン酸化を引き起こすことを見いだした.
3)GPR30は脳血管壁細胞に発現し,重炭酸イオンに応答する
次に筆者らは,生体内におけるGPR30の発現分布を調べることにした.まず抗GPR30抗体を用いて組織染色を行ったが,抗体の特異性が低く,明瞭な像を得ることができなかった(多くのGPCRの配列は細胞膜を貫通する疎水性領域が大部分を占め,抗原となる親水性のアミノ酸配列に乏しいため,特異性の高い抗体を作製することが困難とされている).そこで,GPR30遺伝子座に蛍光タンパク質Venusをコードする遺伝子を挿入したマウスを作製し,全身の臓器を観察すると,脳全体でVenus陽性細胞が発現していた.微小血管壁に発現するtype IV collagenの免疫染色により,Venus陽性細胞が微小血管を取り囲むように存在していることがわかった.さらに,in situ hybridizationおよびVenus陽性細胞の定量PCR解析によって,GPR30は血管壁細胞(血管平滑筋細胞およびペリサイト)に発現していることがわかった.次に,これらのGPR30を発現する血管壁細胞が重炭酸イオンに反応するかどうかを明らかにするため,このマウスから血管平滑筋細胞およびペリサイトを単離し,カルシウムイメージングを行った.GPR30を発現するヘテロのVenusノックインマウス由来の血管壁細胞では重炭酸ナトリウムに反応して細胞内カルシウムが上昇するのに対し,GPR30欠損マウスの血管壁細胞では重炭酸イオンによる細胞内カルシウムの上昇がみられなかったことから,これらの細胞が内在性に発現するGPR30が重炭酸ナトリウムに反応して細胞内カルシウムを上昇させると考えられた.
4)GPR30は血流を制御して脳虚血再灌流障害に関与する
ペリサイトは微小血管において内皮細胞の周りを取り囲むようにして存在する血管壁細胞である.内皮細胞周囲のペリサイトの存在比率は組織によって大きく異なり,中枢神経系や網膜ではペリサイト:内皮細胞=1:1~1:3と生体内で最も高く,骨格筋では1:100程度と報告されている13).ペリサイトの起源およびその機能についてはいまだ議論の余地があるが,血液脳関門(blood-brain barrier:BBB)の維持や血流の調節,血管新生,神経栄養因子の分泌などに関わっていることが知られている.また,脳梗塞の中心的な病態である虚血再灌流障害に関与しているという報告もある14).
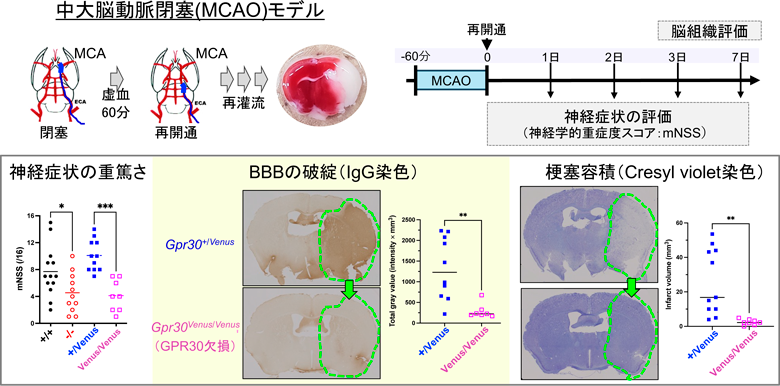
そこでGPR30欠損マウスを用いて一過性局所脳虚血モデル(中大脳動脈閉塞モデル,MCAO)を作製して解析したところ,GPR30欠損マウスでは,野生型マウスと比較して神経症状が軽く,BBBの破綻が軽減し,梗塞容積も小さいことがわかり,虚血再灌流障害が軽減していると考えられた(図3)12).GPR30欠損マウスで虚血再灌流障害が軽減するメカニズムとしては,GPR30欠損マウスでは中大脳動脈の再開通後数時間で神経症状が軽くなっていたことから,血流の回復過程に違いがある可能性が考えられた.そこで中大脳動脈再開通後の動脈血流をMRアンギオグラフィーで,微小循環をドップラー血流計で測定したところ,GPR30欠損マウスでは中大脳動脈の再開通後に動脈血流と微小循環がともに速やかに回復することが明らかになり,GPR30欠損マウスにおける迅速な血流回復が虚血再灌流障害の軽減の一因となっていると考えられた.
これらの一連の結果から,GPR30は血管壁に存在する重炭酸イオン受容体であることが明らかになった.さらに,重炭酸イオンにより惹起されるGPR30は血流を制御し,脳虚血再灌流障害を引き起こすことが明らかになった.
4. 筆者の研究成果からみる今後の脳梗塞治療の展望
現在の脳梗塞治療の柱である,急性期の血栓溶解療法,血管内治療といった再開通治療や脳保護療法(抗酸化剤,抗浮腫療法)をもってしても,6割以上は後遺症を残して制約の多い生活を送っており4),できる限り早い段階からの介入により病態の進行を止めることが喫緊の課題である.
脳梗塞急性期の病態として,まず発症直後から血行動態の急激な変化によって酸塩基平衡や電解質バランスが変化し15),その後,グルタミン酸毒性やカルシウム過負荷,さらに酸化ストレス,炎症が惹起されると考えられている16).この時期に起こるグルタミン酸毒性やカルシウム過負荷を標的とした治療については,いまだ基礎研究の段階である.また,酸化ストレスや炎症に対して抗酸化剤の投与や抗浮腫療法が行われているが,効果は限定的である.
脳梗塞超急性期の酸塩基平衡や電解質バランスが変化する際,GPR30の活性化を抑制することによりいち早い微小循環の回復に寄与できれば,近年の再開通治療の目覚ましい進歩をもってしてもその多くが後遺症を残す脳梗塞に対し,新たな治療法となることが期待される.筆者らは現在,GPR30のアンタゴニストのスクリーニングに着手しており,GPR30に着目した脳梗塞治療への展開を目指している.