1)Noonan症候群類縁疾患の臨床的特徴と原因遺伝子(表1)
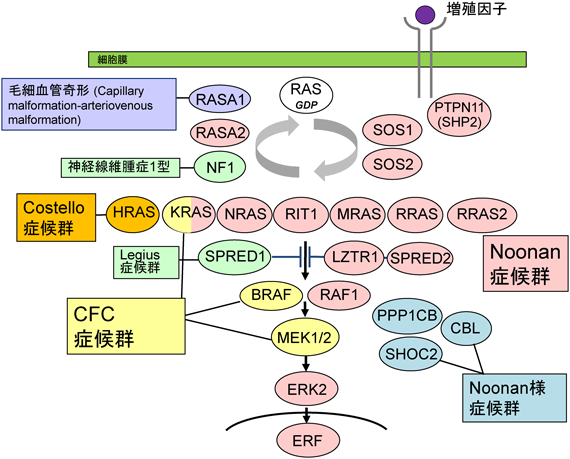
Noonan症候群,Costello症候群,cardio-facio-cutaneous(CFC)症候群は,成長・発達障害,先天性心疾患,肥大型心筋症,さまざまな程度の精神遅滞,特徴的な顔貌,皮膚症状などを示す先天異常症で常染色体顕性/潜性遺伝性疾患である1).Noonan症候群が最も頻度が高く,日本では1万人に1人程度とされている.低身長,先天性心疾患,肥大型心筋症を持ち,精神遅滞は正常から軽度とされている.幼少期の血液増多症やjuvenile myelomonocytic leukemia(JMML)を合併する.Costello症候群,CFC症候群はNoonan症候群に比して頻度はかなり低い.Costello症候群は肥大型心筋症の頻度が高く,手関節の可動性亢進,手掌足底の深いしわ,色黒,乳頭腫などの特徴を有する.Costello症候群の約15%に横紋筋肉腫,神経芽腫,膀胱がんなどを合併する.特に膀胱がんは一般に75歳以上で合併するが,Costello症候群では10代から発生する可能性を有する.CFC症候群は特に幼少時にCostello症候群と類似した症状を有するが,皮膚の症状(黒子,母斑,湿疹など)の合併が多く,精神遅滞もより重度である.Noonan症候群の原因遺伝子としてPTPN11, SOS1, RAF1, RIT1, KRAS, NRAS, SOS2, LZTR1, MAPK1などが同定されている2, 3).Costello症候群の原因はHRAS遺伝子変異である.CFC症候群では,KRAS, BRAF, MAP2K1(MEK1),MAP2K2(MEK2)遺伝子のいずれかに変異が認められる.これらの遺伝子は細胞内RAS/MAPKシグナル伝達経路に存在するためこれらの疾患は包括してRASopathiesと呼ばれる2, 3).類縁疾患としてSHOC2, PPP1CB, CBLの変異を持つNoonan様症候群や,mTOR-AKT経路を活性化するPTPN11変異を持つ多発性黒子を持つ症候群,NF1変異を持つ神経線維腫症1型などがあるが,本稿では詳細はふれない.
表1 Noonan症候群類縁疾患における臨床症状と原因遺伝子| 症候群 | 臨床症状 | 原因遺伝子 |
|---|
| Noonan症候群 | 相対的大頭症,低身長,特異的顔貌,リンパ管異形成,停留精巣,胸郭変形,軽度精神運動発達遅延~正常,血液増殖性疾患
約80%に心血管疾患を合併.肺動脈弁狭窄症,肥大型心筋症心房中隔欠損症,心電図異常 | PTPN11 (40.9%)44) |
| SOS1 (19.3%)44) |
| RAF1 (10.2%)44) |
| RIT1 (~10%) |
| KRAS (2.4%)44) |
| NRAS (0.2%)44) |
| SOS2 |
| MRAS |
| RRAS |
| RRAS2 |
| LZTR1 |
| MAPK1など |
| Costello症候群 | 哺乳障害,体重増加不良,特異的顔貌,低身長,相対的大頭症,カールした毛髪,手掌・足底の深いしわ,精神運動発達遅延,乳頭腫,悪性腫瘍の合併(15%) | HRAS |
| 肥大型心筋症(~60%),先天性心疾患(~44%),肺動脈弁狭窄症(~22%),心房性不整脈(48%) |
| cardio-facio-cutaneous(CFC)症候群 | 特異的顔貌,低身長,皮膚疾患(母斑,黒子,魚鱗癬様皮膚),手掌・足底の深いしわ,精神運動発達遅延,けいれん | BRAF (50%)
MAP2K1/2 (15%)
KRAS (5%>) |
| 約75%に何らかの心疾患を合併.肺動脈弁狭窄症(~45%),肥大型心筋症(~40%),房室中核欠損症 |
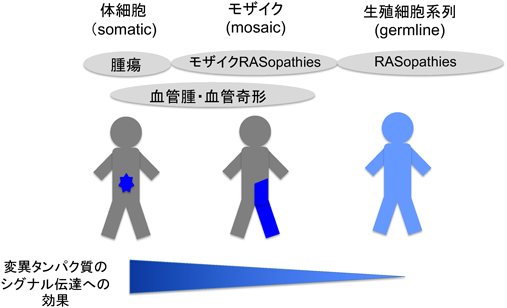
Noonan症候群類縁疾患の原因は生殖細胞系列における遺伝子変異であるが,モザイク状にRASの変異を持つモザイクRASopathiesも多数報告されている.さらに最近,血管奇形の病変部にも体細胞性(somatic)あるいはモザイクのRAS関連遺伝子の変異が同定され4–6),その疾患スペクトラムは広がっている(図1).
2)RASopatihes原因遺伝子同定の経緯と分子メカニズム
2001年にアメリカのMount Sinai病院のTartagliaらのグループが,Noonan症候群の原因遺伝子がSHP-2をコードするPTPN11遺伝子であることを報告した7).アメリカ留学時,神経幹細胞特異的にSHP-2(PTPN11がコードするタンパク質)の機能を低下させるマウスを作製しそのシグナル伝達における影響について研究を行っていた青木は,PTPN11の機能獲得性変異がNoonan症候群を引き起こすことに興味を持った.日本の小児遺伝専門医の先生方にNoonan症候群類縁疾患の患者検体をお送りいただき,新規原因遺伝子同定を目的に候補遺伝子解析を行い,2005年にHRASの生殖細胞系列での変異がCostello症候群の原因であることを発見した8).さらに2006年には当研究室の新堀らがCFC症候群の原因がKRAS, BRAFであることを報告した9).その後,筆者らのグループを含めたさまざまなグループから次々とNoonan症候群類縁疾患の原因としてRAS/MAPKシグナル伝達経路の分子が同定され,筆者らはRAS/MAPK症候群という新しい疾患概念を提唱した(現在はRASopathiesといわれている)2).各疾患に同定されている遺伝子を図2に示した.これらの変異は原則的にはRAS/MAPKシグナル伝達経路を活性化させる変異(がんで同定される変異と重複あり)であり,LZTR1とSPRED2以外は常染色体顕性遺伝形式をとる3).
2014年までのサンガー法による候補遺伝子解析にて,生化学の教科書に記載されている代表的分子(HRAS, KRAS, BRAF, SOS1, RAF1, BRAF, MAP2K1/2)に患者の原因が同定されたが,まだ40%程度が原因不明だった.ちょうどそのころ初めて次世代シークエンサーが東北大学医学部に納入された.筆者らと細胞増殖制御分野の中山啓子先生の研究室で共同して次世代シークエンサーを用いた全エクソーム解析のパイプラインを確立した.そこで原因不明のNoonan症候群類縁疾患患者のサンプルを解析したところ,RIT1変異がNoonan症候群4人に同定された.同定されたRIT1変異は古典型RASと異なり,Switch IIドメインに局在していた.その当時RIT1は神経系のGTPaseであるRIN1との相同性から神経系に発現する分子として解析が行われていたこと,またノックアウトマウスは症状がないがp38MAPKのシグナルと関連するという報告しかなく,RIT1変異がERKを活性するのかは不明で結論には時間を有した.変異を導入した細胞にてELK転写活性が弱いものの増強すること,加齢医学研究所の小椋利彦先生らとの共同研究にてゼブラフィッシュに変異RNAを導入することにより,心臓異常,頭部を含めた骨格異常などが観察されたためNoonan症候群の原因として報告した10).その後RIT1変異陽性Noonan症候群は次々と報告され,現在ではRIT1はNoonan症候群で3番目に頻度の高い遺伝子となり,肺がんを含めたがん研究も行われている.
その後,MRAS, RRAS, RRAS2の変異もRASopathies患者に同定された.非古典型RASで同定された変異の分布はG1ドメインのみならずSwitch IIドメインにわたっている.以上の解析より,ヒト疾患では,古典型RASであるHRAS, KRAS, NRASと非古典型RASであるRIT1, MRAS, RRAS, RRAS2の変異は同様にRASopathiesの原因となることが明らかになった.
3)RASopathiesモデルマウス作製と病態の解明
ある組織にRAS変異を持つマウスはがんの領域でも研究されてきたが,生殖細胞系列において常染色体顕性遺伝(優性遺伝)形式で活性化変異を持つ個体は作製されていなかった.RASopathiesのさまざまな原因遺伝子同定後,活性化変異を持つノックインマウスが作製されてきたが,骨格・心臓の異常が主な表現型であり,血球増加などは報告されているが明らかな自然発がんの報告はない1).筆者らも3疾患のモデルマウス(Braf Q241R変異,Hras G12S変異,Rit1 A57G変異を持つノックインマウス)を作製し報告した11–13).これらのマウスはRIKEN BRCから分与可能である(https://mus.brc.riken.jp/ja/mouse_of_month/nov_2022_mm).
3. 新規原因遺伝子LZTR1とRASのユビキチン化機能と疾患との関係性
1)遺伝子名の由来と機能概念の変遷
LZTR1は,1995年に染色体22q11.2の微細欠失により発症するDiGeorge症候群(22q11.2欠失症候群)の原因遺伝子探索により配列が同定された遺伝子であり,そのアミノ酸配列にロイシンジッパー様モチーフを含むことから「leucine zipper-like transcription regulator 1」と命名された14).当初,その名前が示すように転写因子として核内で機能する可能性が考えられていた.その後,2006年のNacakらの研究によりLZTR1は細胞質のゴルジ体に局在すること,LZTR1はN末端側にKelchドメイン,C末端側に二つのBTB-Backドメインを有する非典型的なBTB-Kelchスーパーファミリー分子である可能性が示された15).一般にBTB-Kelchスーパーファミリーに属する分子は,NRF2-KEAP1経路のKEAP1に代表されるようにユビキチン修飾分子複合体の基質アダプター(足場タンパク質)として機能する16, 17).実際にNacakらの研究では,LZTR1はアポトーシス誘導時に分解されるゴルジ体局在タンパク質であり,転写制御よりも細胞小器官(オルガネラ)の機能維持やタンパク質分解経路に関与する可能性が示された.決定的だったのは,LZTR1がユビキチンリガーゼ複合体の一部(Cul3-RBX1-Kelch複合体)の基質認識サブユニットとして機能することの発見である18).現在では,LZTR1は転写そのものを調節するのではなく,ユビキチン修飾反応を介した翻訳後修飾制御を担う分子と位置づけられている.このため,2024年ごろにleucine zipper like post translational regulator 1と名称が変更された(なお,遺伝子・タンパク質名表記はLZTR1から変更なし).
2)LZTR1遺伝子変異とNoonan症候群
LZTR1遺伝子変異は2015年にNoonan症候群患者で同定された比較的新しいNoonan症候群の原因遺伝子である19).当初は既存のNoonan症候群原因遺伝子同様にヘテロ接合性の常染色体顕性遺伝性の変異として報告されたが,2018年ごろにかけて両アレルの変異による常染色体潜性遺伝性の変異が相次いで報告された20, 21).Johnstonらは,健常な両親から生まれた複数のNoonan症候群患者においてLZTR1の両アレルに病的変異を見いだし,LZTR1遺伝子変異によるNoonan症候群発症には常染色体顕性遺伝だけでなく常染色体潜性遺伝形式も存在することを初めて明らかにした.当研究室の梅木らによる報告においても,常染色体顕性遺伝と常染色体潜性遺伝形式の両タイプのLZTR1遺伝子変異を同定している22).常染色体潜性遺伝形式では,両親は変異の保因者であるがNoonan症候群に特徴的な顔貌異常などの表現型を示さず,患者で二つの変異を有することでNoonan症候群を発症し,その変異はKelchドメインとBTB-Backドメインの両方で同定されている.これらは,ナンセンス変異やフレームシフト変異などLZTR1タンパク質の機能を喪失させる変異(loss-of-function変異)であるとされている.他方,常染色体顕性遺伝形式では,片アレルのみの変異で発症し,特にKelchドメイン上のミスセンス変異が多く報告されており,基質結合部位であるKelchドメインの立体的なリピート構造が損なわれることで標的基質との結合能を失うと考えられている3).
LZTR1変異によるNoonan症候群患者の臨床像はほかの原因遺伝子により発症したNoonan症候群と重なる.出生時から筋緊張低下や哺乳不良がみられることがあり,乳児期以降は発育障害(低身長),特徴的顔貌(眼瞼裂斜下,低位後回転耳,翼状頚など),先天性心疾患(肺動脈弁狭窄や心肥大など)を呈する割合が多い3, 22, 23).これまでの症例報告を勘案すると,常染色体顕性遺伝形式でNoonan症候群を発症する場合には症状が重篤化する傾向にあり,心筋症や乳児白血病の合併,胎児水腫による胎児死亡例も報告されている3, 22, 24).なおグリオブラストーマでLZTR1の体細胞遺伝子変異が報告されたほか,家族性シュワノマトーシス(神経線維腫症の一種)の原因遺伝子としてLZTR1の生殖細胞系列変異が同定された25).
3)LZTR1によるRASサブファミリーのユビキチン修飾制御機構の解明
LZTR1の機能は長年にわたり解明されておらず,その標的基質分子や,Noonan症候群などの疾患発症に関わる分子メカニズムは不明のままであった.LZTR1の機能解析は,2018年末に大きな転換点を迎えることになる.二つのグループから同時にLZTR1はRASサブファミリーのモノユビキチン修飾を制御する分子であることがScience誌に2報同時に掲載された26, 27).Science誌に報告された2グループの研究では,マウスにおけるLztr1ホモ接合性欠失(ノックアウト)では当該マウスは胎生致死であり,Lztr1ヘテロ接合性欠失の場合にはNoonan症候群様表現型として心拡大,顔貌異常を呈した.またLZTR1は古典型RASであるHRAS, KRAS, NRASと結合し,CUL3型ユビキチンリガーゼ複合体の基質アダプターとして機能することでモノユビキチン修飾を促すことを明らかにした.特にRASの170番目のリシン残基へのモノユビキチン化がLZTR1により制御されており,これによりRASの細胞膜局在が減弱することで下流のMAPKシグナル伝達経路の活性レベルが抑制されることを示した.興味深いことに彼らの報告では,LZTR1はRASのタンパク質レベルには影響を及さなかった.
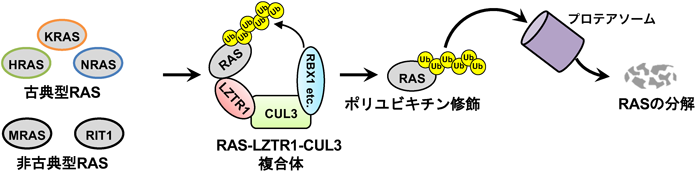
Science誌に掲載された2報とほぼ同時期にCastelらのグループと我々のグループもLZTR1とRASの関係性について報告した28, 29).Castelらのグループは,質量分析装置を用いた相互作用解析によりLZTR1の標的基質として非古典型RASの一つであるRIT1を同定した.一方,我々のグループではLZTR1の標的基質として三つの古典型RAS(HRAS, KRAS, NRAS)と非古典型RASであるMRAS(RRAS3)を報告した.Castelらならびに我々のグループの報告と先行した二つのグループの大きな違いは,LZTR1がRASの分解に関与するか否かである.我々の解析において,LZTR1をノックアウトまたはノックダウンした場合には,RASサブファミリーが細胞内に異常蓄積していく様子が観察された.そこで,LZTR1を過剰発現させたところRASサブファミリーのタンパク質レベルは顕著に低下し,これに呼応してMAPKシグナル伝達経路の活性化レベルも減弱した.そこで,LZTR1存在下または非存在下におけるRASのユビキチン修飾レベルを評価したところ,LZTR1存在下において顕著なRASサブファミリーのポリユビキチン化が認められた.cycloheximide chase assayにより,RASサブファミリーのタンパク質半減期を評価するとLZTR1の存在化ではタンパク質分解が亢進しており,プロテアソーム阻害剤処置によりこの分解促進は解除された.これらの結果より我々は,LZTR1はがん原遺伝子産物RASサブファミリーの分解制御をつかさどるRASキラー分子であると結論づけた(図3).同様に,CastelらのグループにおいてもLZTR1はRIT1の分解制御に関与していることが示されている.また,RIT1 p.M90Iのような特定の変異体ではLZTR1による分解制御を受けなくなり,これによって野生型RIT1と比較してMAPKシグナル伝達経路が相対的に活性化することをCastelらは証明した.我々の検討では,KRAS p.G12Cやp.G12Vまたはp.G13Dやp.G13Sといったさまざまながん種で高頻度に同定されるドライバー変異体においてもLZTR1は分解促進作用を保持していることを明らかにしている29, 30).
LZTR1によるRASサブファミリーのユビキチン修飾制御はCastelらの論文を含め3報がScience誌に記載されるなど大きなインパクトを与えた.一方で,LZTR1はRASのモノユビキチン修飾(局在変化)を制御するのかポリユビキチン修飾(分解誘導)を制御するのか,はこれらの論文が発表された当時は不明であった.しかし,最近のさまざまな追加報告を含めるとLZTR1はRASのポリユビキチン修飾(分解誘導)を制御するとの考え方が主流となっている30–34).
4)LZTR1によるRASサブファミリー以外の標的分子の同定
LZTR1はRASサブファミリー以外にも複数のユビキチン修飾標的が報告されており,その中からいくつか厳選して紹介する.一つ目は,charged multivesicular protein 1B(CHMP1B)である35).CHMP1Bは,endosomal sorting complex required for transport III(ESCRT-III)構成分子の一つであり,核膜の破綻や細胞分裂期後の核小胞再形成,リソソームやエンドソームなどのオルガネラ膜の修復,ならびに小胞輸送などへの関与が指摘されている36).Sewduthらの報告では,LZTR1はCHMP1Bのユビキチン化を制御することによってvascular endothelial growth factor receptor 2(VEGFR2)の小胞輸送を促進しており,LZTR1のヘテロ接合性欠損時にはVEGFR2の小胞輸送が阻害されることで血管新生に障害を及ぼすことが示された.
二つ目はepidermal growth factor receptor(EGFR)とAXL receptor tyrosine kinase(AXL)である33).これら二つはともにチロシンキナーゼ型受容体であり,さまざまな生体内シグナル伝達経路の制御に関与する.Koらのグループは,さまざまな組織由来の細胞株を用いて生体内で普遍的にLZTR1によるユビキチン修飾を受ける標的分子を網羅的に探索し,EGFRとAXLを同定した.より詳細に解析したところ,LZTR1の発現量とEGFR/AXLの発現量は逆相関しており,LZTR1欠損時にはEGFR/AXLのタンパク質半減期は顕著に延長された.いくつかのチロシンキナーゼ型受容体は,リガンド結合後に初期エンドソームに内包され,リソソームとの膜融合後に分解される37–39).そこで,プロテアソーム阻害剤とリソソーム阻害剤を処置して検討を実施すると,EGFR/AXLはプロテアソームを介した分解制御は受けておらず,リソソームを介して分解されることが明らかとなった.さらに,シュワノーマ患者の検体において,LZTR1の生殖細胞系列変異が同定された検体ではEGFRとAXLが有意に発現増加することも示した.Koらの発見により,LZTR1はプロテアソーム経路とリソソーム経路の二つの経路を介してさまざまな生体内分子のプロテオスタシス制御を担うユニークな分子であることが示された.
三つ目は,我々のグループが報告した間接的なSEC31Aのユビキチン修飾の制御である30).我々は,RASサブファミリー以外のLZTR1の標的基質を同定することで,より詳細なLZTR1機能の解析を目指した.抗LZTR1抗体で免疫沈降を実施したサンプルを質量分析装置により解析したところ,複数のLZTR1標的分子候補を同定した.その一つが,Kelch-like protein 12(KLHL12)である.KLHL12は,LZTR1と同様にBTB-Kelchスーパーファミリーに属する分子であり,coat protein complex II(COPII)の構成分子の一つであるSEC31Aのモノユビキチン化を制御する.KLHL12によりSEC31Aがモノユビキチン化するとCOPIIのサイズが増大し,プロコラーゲンを内包できるようになる.これにより,プロコラーゲンは細胞外に輸送・分泌される40–42).LZTR1, KLHL12, SEC31A間のタンパク質相互作用の有無を検討したところ,LZTR1-KLHL12, KLHL12-SEC31Aの結合が認められたがLZTR1とSEC31Aは結合しなかった.一方で,LZTR1ノックダウン時にはSEC31Aのモノユビキチン化レベルが顕著に増加した.この結果より,LZTR1はKLHL12によるSEC31Aのユビキチン化を負に制御しているのではないかと仮説を立てた.実際に,LZTR1の過剰発現時にはKLHL12によるSEC31Aのユビキチン化が抑制された.さらに,LZTR1欠損マウスから採取したmouse embryonic fibroblast(MEF)において,コラーゲンの細胞外輸送の速度を解析したところLZTR1欠損により細胞外へのコラーゲン分泌が顕著に亢進していた.これらの結果より,LZTR1はKLHL12によるSEC31Aのユビキチン化を抑制(プロコラーゲン輸送を負に制御)しており,LZTR1欠損時にはこの抑制が外れることでSEC31Aのユビキチン化亢進を介したプロコラーゲン輸送の促進を引き起こすことが明らかとなった.一般に,BTB-Kelchファミリーはホモまたはヘテロ二量体を形成することで標的基質を捕捉している.LZTR1は,典型的なBTB-Kelchスーパーファミリーとは異なりBTBドメインを二つ有しており,種々のBTB-Kelch分子と多量体を形成することで相手方のBTB-Kelch分子の生理機能を抑制するのではないかと予想される.
5)LZTR1遺伝子変異によるNoonan症候群の発症機序解明
これまでの解析では,LZTR1変異は単純な機能喪失型変異であると考えられていたため遺伝子欠損マウスを解析に用いるのが主流であった.しかしながら遺伝子欠損マウスは常染色体潜性遺伝形式のモデルとしては有用であるが,片アレル変異のみで症状を発現する常染色体顕性遺伝性変異モデルとしては不十分であり,なぜ単一の遺伝子変異のみで症状を発症するのかは明らかとなっていなかった.そこで我々は世界で初めてとなる常染色体顕性遺伝性変異型マウスを作製し,常染色体顕性遺伝性変異によるNoonan症候群発症機序の解明を目指した.作製したマウスは,基質結合部位であるKelchドメインに位置するp.G245R(ヒトp.G248R相当),p.R409C(ヒトp.R412C相当)の2系統の変異マウスである.
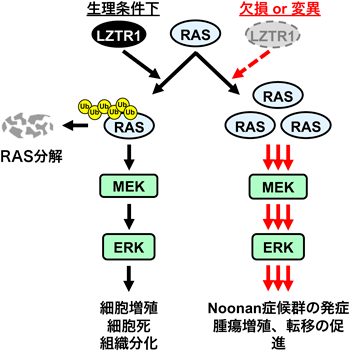
はじめに胎児期のマウスについて解析を実施した.その結果,Lztr1遺伝子欠損マウスと同様にホモ接合性変異マウスは胎生致死であり,ヘテロ接合性変異マウスは出生可能であった.また,野生型マウスとヘテロ接合性変異マウスの出生比率はメンデル比とは大きくかけ離れており,ヘテロ接合性変異マウスには部分的な胎生致死性が示唆された.なお,Lztr1ヘテロ欠損マウスではこのような出生比率の低下は認められていない.そこで,胎生致死性の原因を明らかにするために胎生14.5日,16.5日,18.5日の仔マウスを解析した結果,心臓発生には明らかな変化は認められなかったものの,ヘテロ接合性変異マウスでは軽度浮腫,軽度皮下出血が認められた.さらに,浮腫の原因を解明するために胎仔皮膚のwhole mount stainingによりリンパ管の発生異常を解析したところ,野生型マウスに比べてヘテロ接合性変異マウスではリンパ管の増生異常が認められ,これが胎仔浮腫の原因であることが示唆された.これらの表現型はホモ接合性変異マウスでも認められ,ヘテロ接合性変異マウスに比べてより重篤であった.胎生13.5日の仔マウスよりMEFを採取し,RASサブファミリーの発現量を解析したところ,野生型<ヘテロ接合性変異マウス<ホモ接合性変異マウスの順でRASサブファミリーが増加しており,表現型の重篤度と一致していた.一方で,ヘテロ接合性欠損マウスではRASサブファミリーの発現変動は認められなかったため,ヘテロ接合性変異マウスは単純な機能喪失型変異ではない可能性がこの結果からもうかがえる.
続いて,成体マウスを用いてNoonan症候群様の表現型が認められるかを調査した.その結果,ヘテロ接合性変異マウスは出生時低体重,出生時低身長,特徴的顔貌,心肥大,脾腫,などの表現型を呈した.特徴的顔貌として,眼間開離,短鼻,丸みを帯びた頭蓋骨,などの特徴がmicro-CT解析で示された.心肥大や脾腫は我々がこれまでに作製・解析を実施してきたほかのRASopathiesモデルマウスと共通する特徴的な表現型である12, 13).心臓について解析を進めると,ヘテロ接合性変異マウスの心臓では心筋細胞のサイズが野生型に比べて肥大しており,RASサブファミリーのうち特にMRASやRIT1の発現増加がより顕著であった.心臓サンプルを用いてRAS-seqやプロテオーム解析により,MAPKシグナル伝達経路の活性亢進とmTOR経路の活性低下が示された.そこで,MAPKシグナル伝達経路の構成分子であるMEK1/2の阻害剤であるトラメチニブを4週齢(離乳時点)から12週齢まで混餌投与し,MEK阻害剤の治療効果を検討した.その結果,トラメチニブ投与により心筋細胞のサイズが改善され,心臓重量も野生型と同等レベルまで回復するなど顕著な治療効果が認められた.以上の結果より,ヘテロ接合性変異マウスは生体内でのRASサブファミリーの発現増加を引き起こすことでMAPKシグナル伝達経路を活性化させることで心肥大を引き起こすことが初めて立証された(図4).
ここまでの結果より,ヘテロ接合性変異マウスは単一変異でNoonan症候群様の表現型を呈することが明らかとなったが,ヘテロ欠損マウスとの比較より単純な機能喪失型ではないことが予想された.そこで,LZTR1ノックアウトHEK293(LZTR1-KO)細胞を用いてLZTR1変異体の機能を詳細に解析した.LZTR1-KO細胞において,野生型LZTR1を過剰発現させた場合には古典型RASやMRAS, RIT1などのタンパク質発現は顕著に低下した.他方,常染色体顕性遺伝性の変異体を過剰発現させた際にはこのような変化は認められなかった.同様に,MAPKシグナル伝達経路の活性化レベルを下流転写因子であるETS transcription factor ELK1(ELK1)活性評価系(ルシフェラーゼレポーターアッセイ)により解析したところ,タンパク質レベル同様に野生型ではELK1の転写活性低下が認められたが,常染色体顕性遺伝性の変異体では変化はなかった.これらの結果より,常染色体顕性遺伝性の変異体は野生型LZTR1が有するRASサブファミリーのプロテオスタシス制御能を喪失していることがわかる.続いて,常染色体顕性遺伝性の変異体が野生型LZTR1に対してドミナントネガティブ体として機能するかを検討した.LZTR1-KO細胞に一定量の野生型LZTR1を発現させ,常染色体顕性遺伝性の変異体を共発現させた際のELK1転写活性レベルの変化をみると,常染色体顕性遺伝性の変異体の量依存的に野生型によるELK1転写活性の低下が抑制された.また,in vitro翻訳タンパク質を用いて野生型LZTR1と常染色体顕性遺伝性の変異体の結合評価を実施したところ,野生型LZTR1のホモ二量体形成は低濃度の常染色体顕性遺伝性変異体添加により阻害されることを見いだした.すなわち,LZTR1常染色体顕性遺伝性の変異体はRAS分解能を喪失しているのに加えて,野生型LZTR1の二量体形成を阻害することでドミナントネガティブ体としても機能しており,これによって単一遺伝子変異によりNoonan症候群様の症状が発現することが強く示唆された32).