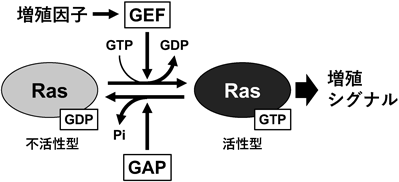
他の稿においても記述されていることと思うが,Rasファミリーを代表とする低分子量Gタンパク質(small GTPases)の多くは,GTP(グアノシン三リン酸)結合状態で活性化し,GDP(グアノシン二リン酸)結合状態で不活性化する1).これらは細胞内シグナル伝達における分子スイッチとして機能し,細胞増殖,分化,細胞運動,膜輸送などの多様な生理機能を制御する2).Rasファミリーは,GTP/GDPの交換およびGTPの加水分解を介して活性が制御されるが,これらの過程は,GEF(guanine nucleotide exchange factor)やGAP(GTP-hydrolysis activating protein)による厳密な調節を受ける(図1).たとえばRasの場合,Sos(Son of Sevenless)やRasGRFがGEFとしてRasのGDP-GTP交換反応を促進する3).GTP結合型のRasは下流のシグナル分子(エフェクター)を活性化し,細胞増殖シグナルを刺激するが,過剰な活性化を防ぐためにGTPを加水分解して元の不活性型であるGDP結合型に戻る.この際,p120GAPやNF1(neurofibromin 1)などのGAPがGTP加水分解を促進する3).また,Rho, Rab, Arfファミリーなどは,細胞骨格制御や膜輸送などのRasとは異なる細胞機能を担い,それぞれ特異的な制御機構を持つ.
2. κB-Rasの同定とκB-Rasタンパク質の性質と機能
κB-Rasは,NF-κBの制御因子であるIκBαをbaitとした酵母two hybrid screeningを行ったGhoshの研究グループによりクローニングされた4).NF-κBは,免疫応答や炎症のみならず,発生やアポトーシスの調節,腫瘍の発生や悪性化に関与するさまざまな遺伝子の発現を活性化する転写因子のファミリーとして知られる5).哺乳類のNF-κBには5種類のサブユニット,p50, p52, p65/RelA, c-Rel,およびRelBが存在し,これらがヘテロ二量体やホモ二量体を形成して機能する.ここでは代表的なp65/RelAとp50のヘテロ二量体をNF-κBとして述べる.未刺激の細胞内では,NF-κBは調節分子であるIκBファミリーと不活性型の複合体を形成し,細胞質に局在している.たとえば,p65/RelA·p50ヘテロ二量体は,IκBαやIκBβと相互作用する.サイトカインや細菌由来のリポ多糖(LPS)などの刺激によりIκBキナーゼ(IKK)が活性化されると,IKKはIκBをリン酸化し,IκBのユビキチン・プロテアソーム系を介してタンパク質分解を促進する.この結果,放出されたNF-κBは核に移行し,標的遺伝子のプロモーター・エンハンサー配列に結合して標的遺伝子の発現を促進することがわかっている.Ghoshのグループは,κB-RasがIκB,特にIκBβを安定化させ,NF-κBの活性化を抑制する分子モデルを提唱したが,その詳細は不明だった4, 6, 7).
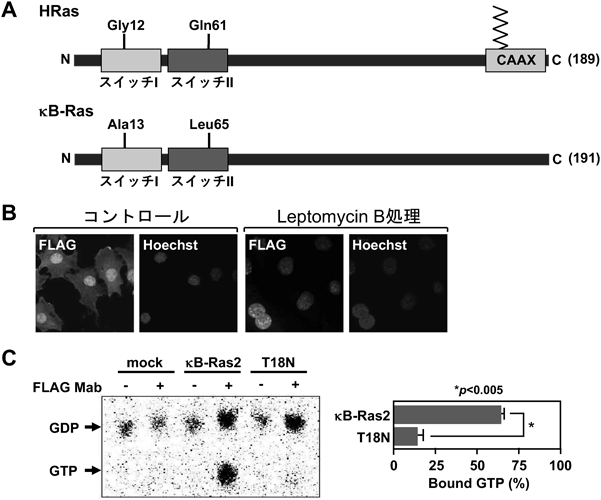
κB-Rasには,非常に高い相同性を示すκB-Ras1とκB-Ras2という二つの分子種が存在する.κB-Rasは,ヒトやマウスだけでなく,ショウジョウバエやニジマスなど,多くの生物種にみられる4, 8).κB-Rasは,他のRasファミリーなどの低分子量Gタンパク質と同様に,よく保存されたスイッチIおよびスイッチII領域を有する.スイッチIおよびスイッチII領域は,GDPやGTPといったグアニンヌクレオチドと結合し,下流に位置するシグナル分子(エフェクター)や活性調節分子であるGEFやGAPとの相互作用に機能する.また,この領域にはほかのRasファミリーと同様に,グアニンヌクレオチドと結合する際に重要なMg2+との相互作用に関与するアミノ酸残基(Thr-18およびThr-36)も保存されている.一方で,κB-Rasには,他のRasファミリーと比較していくつかの構造的特徴が存在する(図2A).第一に,κB-Rasは,他のRasファミリーがGly残基を有するN末端側から13番目の位置にAlaまたはLeuを有し,65番目のGlnの位置にはLeuを持つ.これらのアミノ酸残基はGTP加水分解活性を示すのに重要であり,Rasファミリーの機能が結合するグアニンヌクレオチドにより調節されるため,κB-Rasが細胞内でどのようなグアニンヌクレオチド結合状態にあるかは重要な問題であった.第二にκB-Rasは,多くのRasファミリーが持つCAAXモチーフと呼ばれるC末端側の膜結合配列(Aは任意の疎水性アミノ酸,Xは任意のアミノ酸)を欠いており,κB-Rasの細胞内局在は不明だった.
まず我々は,κB-Rasがどのような性質の低分子量Gタンパク質かを明らかにすることを試みた9).κB-Ras1とκB-Ras2を比較すると,κB-Ras2の方が多くの組織や細胞種で発現しているため,主な解析ではκB-Ras2について調べた.まず,HeLa細胞やNIH-3T3細胞を用いてκB-Ras2の細胞内局在を免疫染色により調べた結果,κB-Ras2は核内および細胞質中に局在していることが観察された.さらに,CRM1阻害剤であるleptomycin Bで細胞を処理したところ,細胞質に存在したκB-Ras2は消失し,核内のみに局在する結果が得られた(図2B).CRM1は,核外移行シグナル(nuclear-export signal:NES)を持つタンパク質やその複合体を認識して核から細胞質に輸送する機能を担う10, 11).したがってκB-Ras2の局在はCRM1により制御されていることが示唆された.しかし,κB-Ras2の構造中にNESあるいはそれに類似したアミノ酸配列は見いだされなかった.このことから,κB-Rasの核局在は,おそらくκB-Rasの相互作用分子によって制御されると考えられた.
次に我々は,細胞内におけるκB-Rasのグアニンヌクレオチドの結合状態を調べた.多くのRasファミリーが血清に含まれる細胞増殖因子により活性化されるため,無血清培地で培養した細胞中のκB-Ras2を免疫沈降法により精製し,κB-Ras2が結合しているグアニンヌクレオチドを薄層クロマトグラフィーで分離,検出した.その結果,未刺激の細胞においてもκB-Ras2は半分以上がGTP結合型で存在することがわかった(図2C).我々は,κB-Rasのグアニンヌクレオチドとの結合が局在や活性を制御する可能性を検討するため,κB-Ras2の18番目のThr残基をAsnに変異したκB-Ras2(T18N)変異体を作製した.このκB-Ras2(T18N)変異体を用いて結合するグアニンヌクレオチドを調べたところ,GTP結合型はほとんど消失し,GDP結合型のみが存在することが確認された.さらに,κB-Ras2(T18N)変異体の細胞内局在を調べた結果,野生型κB-Rasと異なり,T18N変異体は細胞質のみに局在することが観察された.以上の結果から,κB-Rasの細胞内局在は,GDP結合型は細胞質に,GTP結合型が核内にそれぞれ移行することが示唆された.
κB-Rasと類似して,結合するグアニンヌクレオチドにより細胞内局在を変化させる低分子量Gタンパク質としてRanが知られている.RanもGDP結合型は細胞質に局在し,GTP結合型は核内に局在する.しかし,両者のGTP結合型の局在様式はかなり異なる.Ranの場合,核膜孔に存在するタンパク質複合体に含まれ,タンパク質やタンパク質・RNA複合体などの輸送を制御する12).一方で,κB-Rasは染色体上に存在することが示唆されている.κB-Rasの核内での機能は依然として不明であるが,GhoshのグループがκB-Rasを転写因子NF-κBの活性制御に関わる分子として見いだしたことを踏まえると,κB-Rasの核内での役割はきわめて重要であると考えられる.
3. κB-Rasによる転写因子NF-κBの活性制御機構
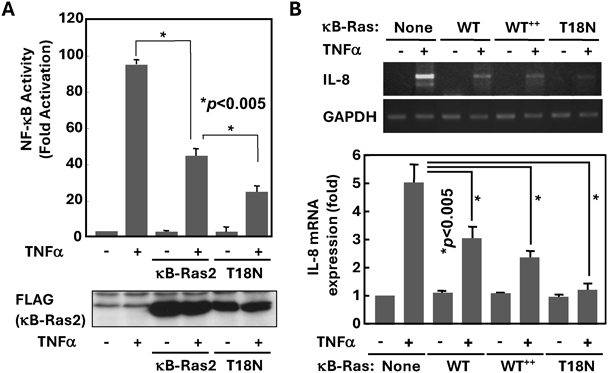
そこで我々は,κB-Rasがサイトカイン刺激によるNF-κBの活性化を抑制する分子機構の解明を目指した9).はじめに,HEK293T細胞を用いたリポーター遺伝子解析によってNF-κBの活性化を調べ,強制発現させたκB-Ras2およびT18N変異体の効果を解析した.野生型κB-Ras2の強制発現は,TNFα(tumor necrosis factor α)刺激によるNF-κB活性化を顕著に抑制した(図3A).さらに,κB-Ras2(T18N)変異体は,野生型κB-Ras2よりもNF-κB活性化に対してより強力な抑制効果を示したことから,GDP結合型のκB-Ras2がNF-κB活性化を強く抑制する可能性が示唆された.同様の結果は,野生型κB-Ras2またはκB-Ras2(T18N)変異体を強制発現させたHEK293T細胞を用いたIL-8 mRNAのRT-PCR解析でも観察された(図3B).
Ghoshらの報告によれば,κB-Ras2はIκBファミリーの特にIκBβの分解を抑制することによりNF-κBの活性化を阻害する分子機構が提唱されている.実際,我々もκB-Ras2がIκBβの分解を抑制することを観察したが,その効果は野生型κB-Ras2とT18N変異体の間で差はなく,リポーター遺伝子解析やRT-PCRの結果とは一致しなかった.IκBβの安定化がNF-κBを阻害する主なメカニズムであるならば,κB-Ras2はサイトカイン刺激によるNF-κBの核移行を抑制するはずである.この可能性を検証するため,NF-κBの核移行およびDNA結合活性に及ぼすκB-Ras2の影響をゲルシフト解析により評価した.その結果,TNFα刺激はp65/RelAのDNA結合活性を亢進したが,κB-Ras2およびT18N変異体のいずれもTNFα刺激によるNF-κBの核移行やDNA結合活性の亢進に影響を及ぼさなかった.この結果は,Ghoshらが提唱するκB-Ras2によるIκBβの安定化がNF-κBを阻害するというモデルでは不十分であることを強く示唆した.
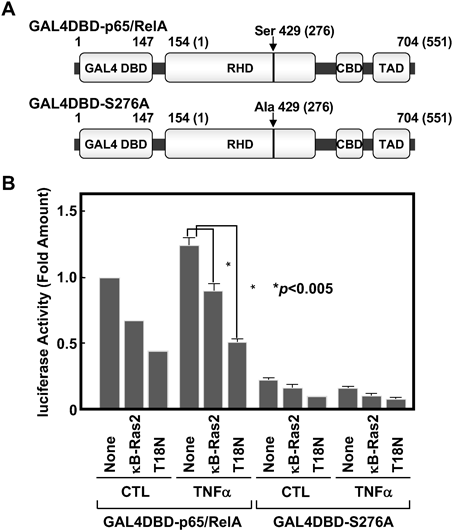
そこで我々は,GAL4DBD-p65/RelAと名づけた,p65/RelAのN末端にGAL4転写因子のDNA結合ドメインを融合したタンパク質の発現プラスミドを作製し,NF-κBの転写活性化に対するκB-Ras2の効果を調べた(図4A).GAL4DBD-p65/RelAの発現ベクターと,GAL4応答配列を有するルシフェラーゼ遺伝子を用いたリポーター遺伝子実験を行ったところ,図4Bに示すように,GAL4DBD-p65/RelAは高い転写活性を示し,TNFα刺激によってさらに活性が強化された.しかし,この転写活性はκB-Ras2およびκB-Ras2(T18N)変異体により著しく阻害された.特に,T18N変異体は野生型よりも強力に抑制効果を示した.p65/RelAが転写活性化されるためには,276番目のSer(Ser-276)のリン酸化が重要であることが報告されている.p65/RelAのSer-276をリン酸化するタンパク質キナーゼとしては,cAMP依存性タンパク質キナーゼ(PKA)やmitogen- and stress-activated protein kinase 1/2(MSK1/2)が知られており13, 14),このSer残基に変異を導入するとGAL4DBD-p65/RelAの転写活性化能は消失する(図4B).我々は,κB-Ras2がp65/RelAに直接結合し,PKAによるp65/RelAのリン酸化を抑制することを明らかにした9).さらに,κB-Ras2によるp65/RelAのリン酸化に対する阻害効果は,T18N変異体でより強いことが示された.
p65/RelAのSer-276のリン酸化は,転写活性化因子p300/CBPとNF-κBの相互作用を引き起こす重要な要因であることが知られている15).これまでの実験結果を踏まえ,我々は研究をさらに進めた結果,κB-Rasがp65/RelAのリン酸化を抑制し,その結果としてNF-κBとp300/CBPの相互作用を阻害することによりNF-κBの活性化を抑制するという新しい分子機構のモデルを提唱した.
4. マウスおよびヒトのがんにおいてκB-Rasはがん抑制シグナル分子として機能する
Oeckinghausらは,κB-Rasの生理機能を解明する目的で,κB-Ras1およびκB-Ras2のダブル欠損マウスを作製した16).この遺伝子欠損マウスは生後数時間で致死に至る状態だったが,TNF受容体1型のノックアウトマウスと交配することにより,その表現型は消失し,出生後も生存可能となった.したがって,κB-Rasのダブル欠損による致死は,TNFαシグナルに依存することが明らかになった.Oeckinghausらは,κB-Rasの発現を欠失した細胞において,NF-κBの転写活性が異常に亢進し,それに伴って炎症性サイトカインの発現が増加することを確認した.このことから,κB-RasはNF-κBの負の調節因子として機能し,過剰な炎症応答を抑制していることがin vivoのレベルでも証明された.さらに彼らは,κB-Rasの新規結合分子として低分子量Gタンパク質の一つであるRalの制御因子であるRalGAPを同定した.κB-Rasが欠損した条件下では,Ralのシグナル経路にも影響がおよび,RalAおよびRalBのGTP結合型が蓄積することが明らかとなった.Ralシグナル経路はがん細胞の性質の一つである足場非依存性増殖能を促進する役割を有し,がん細胞の増殖能と密接に関連している.Oeckinghausらは,軟寒天培地中のコロニー形成能に基づき,がん細胞の足場非依存性増殖能を評価した.その結果,κB-Ras欠損細胞では足場非依存性増殖能が著しく亢進しており,これが腫瘍形成の促進に寄与していることを示した.また,κB-Rasの発現が低下したがん細胞に対し,NF-κBまたはRal経路の阻害剤を使用することで細胞増殖や炎症反応を抑制できることが明らかになった.これにより,κB-Rasの欠損が腫瘍の進行に寄与するメカニズムがNF-κBとRal経路の双方を介していることが示唆された.
さらに,多くのヒト腫瘍組織ではκB-Rasの発現が低下しており,特に悪性度の高い腫瘍において顕著であることが報告されている.これらの腫瘍細胞に外因的にκB-Rasを発現させると,NF-κBおよびRal経路の活性が低下し,軟寒天培地中でコロニー形成能が抑制された.OeckinghausラボのBeelらは,膵臓管状腺がん(PDAC)の発生および進行におけるκB-Rasタンパク質とRal GTPaseの機能を調べた17).この研究では,PDAC組織切片においてκB-Rasの発現低下が頻繁に観察され,その低下ががんの進行度と高く相関することが明らかになった.
また,KRas(G12D)変異を有するマウスモデルを用いた実験により,κB-Rasの欠損が腫瘍発生を加速し,マウスの生存期間を短縮させることが示された.さらに,κB-Ras遺伝子の欠損は,腫瘍発生初期だけでなく,がん進行中における腺房細胞から管状細胞への形質転換(腺房–導管異形成)を促進することが示された.腺房–導管異形成とは,膵臓の慢性炎症において腺房細胞が脱分化を繰り返す中で,一部の細胞が脱分化状態を持続することで生じる形態変化である.κB-Rasの欠損は,細胞増殖だけでなく浸潤にも影響を及ぼすことが示唆された.また,κB-Rasタンパク質は膵炎後の腺房細胞の再生にも必要であり,細胞が環境の変化やストレスに応じて適応する過程で重要な役割を果たすことが示唆されている.分子レベルでは,Ral GTPaseの活性化およびSox9の発現上昇が観察され,これらが腺房–導管異形成を促進する新たなRalシグナル伝達の役割として特定された.以上の結果からBeelらは,κB-Rasが腫瘍抑制因子として機能し,Ralシグナル伝達の制御喪失がPDACのリスク要因となる可能性を示唆した.
Postlerらは多がん種解析を行い,その結果,ヒトのがんにおいて,κB-Ras1とκB-Ras2の発現が同時に減少することや,Ras変異とκB-Rasの発現現象が同時に起こることは非常にまれであることを明らかにした18).一方で,κB-Ras2とは異なり,κB-Ras1の発現低下は,いくつかのヒトがんの予後悪化と関連していることが示された.
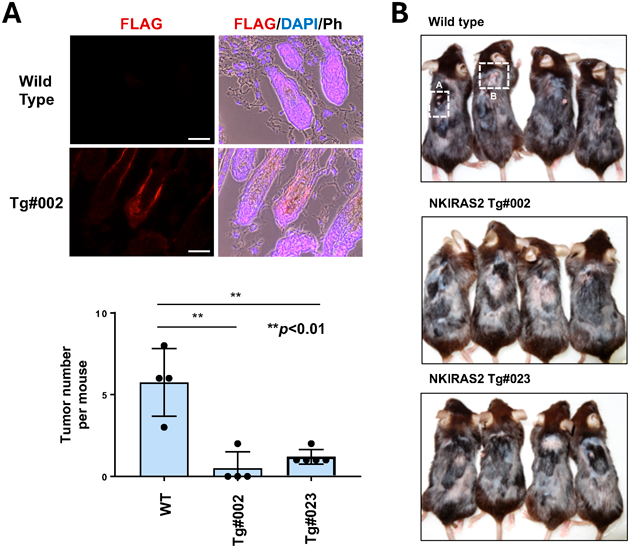
我々の研究グループも,マウスモデルを用いてκB-Rasの生理機能を解析するため,κB-Ras2を強制発現するトランスジェニックマウス(Tgマウス)の作製を試みた.はじめに,κB-Ras2を全身で発現するTgマウスの作製を試みたが,目的のマウスは得られなかった.この原因は不明であるが,細胞種によっては,κB-Rasが細胞増殖を抑制することが原因である可能性がある.そこで,κB-Ras2が皮膚の発達や腫瘍形成に与える影響を調べるため,K15(keratin 15)遺伝子プロモーターを用いてκB-Ras2を強制発現させるTgマウス(K15-NKIRas2-Tg)を作製した19).K15は,皮膚毛包中のfollicle bulgeと呼ばれる膨らんだ構造体に存在する皮膚幹細胞のマーカーとして知られている.FLAGタグを付加したκB-Ras2のcDNAをK15プロモーターの下流に挿入したプラスミドを作製し,そのDNA断片を受精マウス卵母細胞に注入した.その結果,二系列のTgマウス(K15-NKIRas2-Tg#002およびK15-NKIRas2-Tg#023)が得られた.これらのマウス新生仔から皮膚切片を調製し,Tgマウスおよび野生型マウスの皮膚におけるκB-Ras2の発現を調べた.マウスの内在性κB-Ras2(Nkiras2)は,ユビキタスに発現しているが,Tgマウス#002と#023では,毛包におけるκB-Ras2の発現強度が高いことが観察された.また,遺伝子導入したκB-Ras2のアミノ末端にはFLAGタグが付加されているため,抗FLAG抗体を用いてκB-Ras2の外因性発現を検出した.その結果,図5Aに示すように,Tgマウスのfollicle bulgeでFLAG-κB-Ras2の発現が特異的に検出された.
次に,我々はK10, K14, K15,フィラグリンなどの皮膚マーカーに対する免疫蛍光分析を行い,follicle bulgeにおけるκB-Ras2の強制発現が皮膚の発達に及ぼす影響を調べた.マウスの体毛は,成長期(anagen),退行期(catagen),休止期(telogen)の順で移行する毛周期に従って発育することが知られている20).そこで,新生仔(出生後1日または2日),anagen(出生後5週間),telogen(出生後7週間)の3段階で各種皮膚マーカーの発現を解析した.K10は,皮膚表皮のスピノサム層および顆粒層のマーカー,K14は表皮基底層のマーカー,フィラグリンは表皮の顆粒細胞で発現し皮膚のバリア機能に必須なタンパク質である.これらの解析結果から,κB-Ras2の強制発現がマウス皮膚の発達に顕著な影響を及ばさないことがわかった.
続いて,毛包内のfollicle bulgeで強制発現したκB-Ras2がDMBA/TPA処理による化学発がんに与える影響を調べた.マウスの皮膚に発がん性物質であるDMBA(7,12- dimethylbenz[a]anthracene)を塗布することによりHRas遺伝子にがん変異を誘発し(イニシエーション),その後に炎症誘発物質であるTPA(12-O-tetradecanoylphorbol-13-acetate)を数か月間繰り返し塗布することにより良性腫瘍(パピローマ)が形成される(プロモーション)ことが知られている21).図5Bに示すように,野生型マウスではDMBA/TPA処理後に皮膚に腫瘍の形成が観察されたが,κB-Ras2 Tgマウスでは腫瘍の数が顕著に少なかった.この結果は,follicle bulgeにおける皮膚幹細胞でのκB-Ras2の強制発現が,DMBA/TPAが誘発する化学発がんを抑制したことを示唆している.さらに,腫瘍が発生した野生型マウスとκB-Ras2 Tgマウスの皮膚からパラフィン切片を作製し,HE染色を行った.その結果,両者の腫瘍に大きな違いはみられなかった.これらの結果から,K15プロモーターによる皮膚幹細胞でのκB-Ras2の強制発現が,DMBA/TPA処理による細胞形質転換とパピローマの発症を抑制することが示唆された.
5. κB-Rasによるマウス線維芽細胞の形質転換促進効果
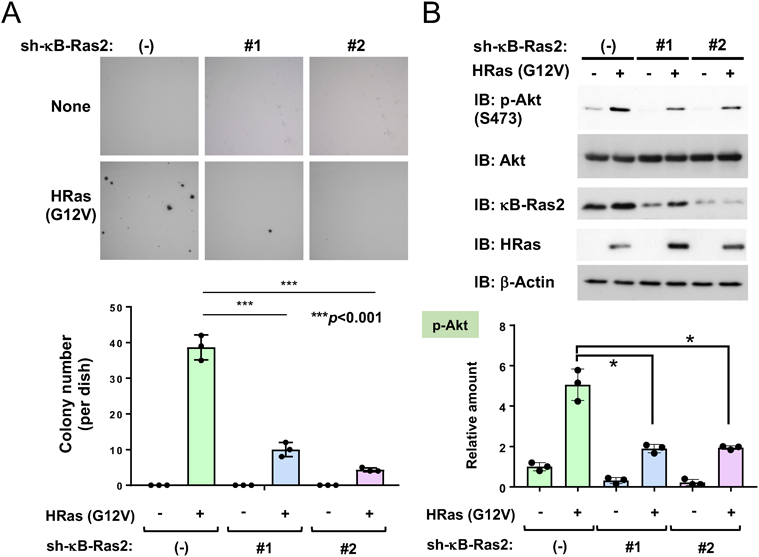
いくつかのがん症例におけるカプラン・マイヤー曲線の解析結果では,腫瘍の予後とκB-Ras1/2の発現レベルの相関関係には依然として議論の余地があることが示されている(NKIRas1:https://www.proteinatlas.org/ENSG00000197885-NKIRAS1/pathology ; NKIRas2:https://www. proteinatlas.org/ENSG00000168256-NKIRAS2/pathology).そこで我々は,細胞がん化(形質転換)とκB-Ras発現の関係についてさらなる洞察を得るため,がん化型Ras変異体[HRas(G12V)]によるマウス線維芽細胞NIH-3T3の形質転換におけるκB-Rasの機能を解析した.NIH-3T3細胞では,内在性κB-Ras2が発現している一方,内在性κB-Ras1の発現はイムノブロットやRT-PCRでは確認されなかった.そこで,我々はκB-Ras2を標的とする低分子ヘアピン型RNA(shRNA)を発現できるレトロウイルスベクターを作製した.これらのshRNAレトロウイルスを用いて,κB-Ras2のノックダウンがHRas(G12V)による形質転換に与える影響を解析した.HRas(G12V)をNIH-3T3細胞に発現させた結果,接触阻止抵抗性や足場非依存性増殖能といったがん細胞特有の性質が獲得された.さらに,軟寒天培地を用いたコロニー形成実験では,HRas(G12V)がコロニー形成を促進することが観察された.一方で,κB-Ras2のノックダウンにより,HRas(G12V)による形質転換が大幅に抑制された(図6A).この結果は,上述のマウスモデルで得られた結果とは対照的であった.
次に,κB-Ras2のノックダウンがERK, JNK, Aktの活性化に及ぼす影響を解析した.Ras依存的な発がんにはこれらのシグナル経路が関与していることが知られているが,興味深いことに,κB-Ras2のノックダウンは,Ras依存的なAktのSer473リン酸化を効果的に抑制した.このAktリン酸化の抑制は,形質転換していない細胞でも観察された(図6B).一方で,κB-Ras2のノックダウンはERKやJNKの活性化を抑制しなかった.特に,ERKの活性化はHRas(G12V)の発現に関係なく,κB-Ras2のノックダウンによってむしろ増強された.これらの結果から,κB-Ras2はマウス線維芽細胞におけるRasシグナル依存的な細胞がん化に必要である一方,Rasの下流シグナルに対しては複雑に影響することが明らかになった.
6. 古典的Rasによる発がんシグナルとκB-Rasシグナルのクロストーク
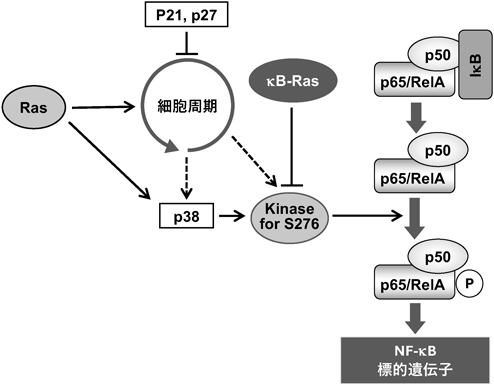
Rasファミリーは,細胞の増殖やがん化だけでなく,転移・浸潤や血管新生といったがんの悪性化にも深く関与している.また,がん細胞の転移・浸潤や腫瘍周辺の血管新生にはNF-κBも関与しており,これがNF-κBを抗がん剤の標的分子の一つとして考える理由となっている.我々は,がん細胞におけるRasシグナルのNF-κB活性化への影響を解析し,HRas(G12V)変異体がTNFα刺激によるNF-κBの活性化を増強することを見いだした22).この活性化増強効果は,KRasのG12V変異体だけでなく,最近報告されたK117N変異体やA146T変異体を発現させた際にも観察された.また,RasシグナルによるNF-κB活性化の増強効果は,細胞周期の進行に依存することが判明した.サイクリン依存性タンパク質キナーゼ(CDK)の阻害タンパク質であるp21Cip1やp27Kip1,さらにがん抑制遺伝子産物p53を活性化するp19Arfの強制発現は細胞周期を停止させ,RasシグナルによるNF-κB活性化の増強を顕著に抑制した.加えて,このNF-κB活性化の増強効果は,タンパク質キナーゼp38阻害剤であるSB203580によって顕著に抑制された.しかし,HRas(G12V)は,TNFα刺激によるIκBαの分解やNF-κBの核内移行には影響を与えなかった.
さらに,Rasの下流でp38を介して活性化されたタンパク質キナーゼMSK1/2がNF-κBのp65/RelAサブユニットにおけるSer-276をリン酸化し,NF-κBの転写活性を増大させるメカニズムが解明された(図7).このp65/RelAサブユニットにおけるSer-276のリン酸化は,大腸がんの腫瘍部位で顕著に亢進しており,新規の腫瘍マーカーになる可能性が示された.Rasによるp38活性化の分子機構は未解明であるが,我々の知見は,p38が細胞周期依存的にG2/M期で活性化されるとの報告と一致した23).また,p65/RelAサブユニットにおけるSer-276のリン酸化は,κB-RasがNF-κB活性化を阻害する際の標的でもあるため9),NF-κBの活性調節を介して古典的Rasの発がんシグナルとκB-Rasの機能的なクロストークががんの悪性化や予後に関与していることが示唆された.
7. κB-Rasとエフェクターの相互作用とシグナル伝達系
低分子量Gタンパク質はエフェクターと相互作用し,その活性を制御することによりシグナル分子として機能する.RasやRac, Rhoなど多くの低分子量Gタンパク質は,GTP結合型で活性型としてエフェクターと結合する.一方,Arf6のようにGDP結合型でRacGEFのKalirinと相互作用し,Racシグナルを調節する例も報告されている24).我々は,κB-Rasがp65/RelAのSer-276リン酸化を抑制することによりNF-κBの活性化を阻害するモデルを提唱しているが,この阻害機構にはp65/RelAとκB-Rasの相互作用が必要である.in vitroプルダウンアッセイの結果,p65/RelAはκB-Rasと直接結合することが確認され,その結合強度はGDP結合型のT18N変異体でより強いことが示された.このことから,p65/RelAとκB-Rasの相互作用はGDP結合型で強固であることが示唆された.
最近,RascheらによりκB-RasとRalGAPの複合体に関する解析結果が報告された25).κB-Rasタンパク質は非常に低いGTP加水分解活性を持ち,GDPおよびGTPとの結合親和性もきわめて低い「ファストサイクリング」GTPaseとして機能することが明らかになった.細胞内では主にGTP結合型で存在することが確認された.また,κB-RasとRalGAPa2の相互作用は,ヌクレオチドの結合状態やスイッチ領域の構造に依存せず,直接結合することが示された.
一方で,κB-Rasの細胞内局在は,結合するグアニンヌクレオチドにより変化するため,細胞内でのエフェクターとの相互作用が間接的にGDPやGTPの結合状態によって制御されることが示唆されている.κB-Rasの活性調節機構,特にGDP-GTP交換反応やGTPの加水分解をサポートするGEFやGAPが存在するかどうかは,いまだ不明である.我々もκB-Ras複合体を精製し,κB-Rasの相互作用分子をいくつか同定している(未発表).これらの相互作用分子の中には,さまざまな低分子量Gタンパク質と結合するsmgGDSが含まれる(未発表).smgGDSは,ある種の低分子量Gタンパク質に対してGEFとして機能することが報告されているが,smgGDSがκB-Rasの活性制御に関与しているかどうかは明らかではない26).κB-Rasと相互作用するsmgGDSの役割については,κB-Rasの核と細胞質間のシャトリングを制御する分子機構と併せて,今後解明が必要な課題といえる.
また,Rascheらは,腫瘍患者でまれにκB-Ras1およびκB-Ras2遺伝子に変異が生じることを報告している25).これらの変異の多くはヌクレオチド結合部位やスイッチ領域に位置し,κB-Rasの機能やRalGAPとの相互作用に影響を及ぼす可能性がある.
κB-RasがNF-κBを阻害する分子機構については,ある程度の結論が得られた一方で,細胞増殖や細胞がん化におけるκB-Rasの具体的な機能については未解明な部分が多く残されている.特に,がん細胞への形質転換や細胞増殖を促進または抑制する場合のκB-Rasの細胞内局在に関する知見は不足している.Oeckinghausらは,RalGAPをκB-Rasのエフェクターとして同定し,κB-RasがRalシグナルを負に制御することを報告しているが,Ralの下流シグナルに関する具体的な解析は行われていない.複数の研究グループにより,RalがRasの下流でJNKの活性を制御し,細胞増殖を調節することが示されているが,我々の研究では,κB-RasのノックダウンがRas変異体によるJNK活性化には影響を与えないことを確認している27).発がんにおけるκB-Rasの役割を解明するには,κB-Rasタンパク質複合体の解析や細胞内局在の調節機構の詳細な研究が今後の課題である.
謝辞Acknowledgments
本稿を書く機会をいただいた山内淳司先生,加藤裕教先生に深くお礼申し上げます.同時に本稿の推敲や全体の組み立てなど,多大なるご助力をいただいた多胡めぐみ先生に心よりお礼申し上げます.
引用文献References
1) Kaziro, Y., Itoh, H., Kozasa, T., Nakafuku, M., & Satoh, T. (1991) Structure and function of signal-transducing GTP-binding proteins. Annu. Rev. Biochem., 60, 349–400.
2) Matozaki, T., Nakanishi, H., & Takai, Y. (2000) Small G-protein networks: Their crosstalk and signal cascades. Cell. Signal., 12, 515–524.
3) Karnoub, A.E. & Weinberg, R.A. (2008) Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol., 9, 517–531.
4) Fenwick, C., Na, S.Y., Voll, R.E., Zhong, H., Im, S.Y., Lee, J.W., & Ghosh, S. (2000) A subclass of Ras proteins that regulate the degradation of IκB. Science, 287, 869–873.
5) Karin, M. (1999) How NF-κB is activated: The role of the IκB kinase (IKK) complex. Oncogene, 18, 6867–6874.
6) Chen, Y., Wu, J., & Ghosh, G. (2003) κB-Ras binds to the unique insert within the ankyrin repeat domain of IκBβ and regulates cytoplasmic retention of IκBβ · NF-κB complexes. J. Biol. Chem., 278, 23101–23106.
7) Chen, Y., Vallee, S., Wu, J., Vu, D., Sondek, J., & Ghosh, G. (2004) Inhibition of NF-κB activity by IκBβ in association with κB-Ras. Mol. Cell. Biol., 24, 3048–3056.
8) Sarais, F., Rebl, H., Verleih, M., Ostermann, S., Krasnov, A., Köllner, B., Goldammer, T., & Rebl, A. (2020) Characterisation of the teleostean κB-Ras family: The two members NKIRAS1 and NKIRAS2 from rainbow trout influence the activity of NF-κB in opposite ways. Fish Shellfish Immunol., 106, 1004–1013.
9) Tago, K., Funakoshi-Tago, M., Sakinawa, M., Mizuno, N., & Itoh, H. (2010) κB-Ras is a nuclear-cytoplasmic small GTPase that inhibits NF-κB activation through the suppression of transcriptional activation of p65/RelA. J. Biol. Chem., 285, 30622–30633.
10) Wen, W., Meinkoth, J.L., Tsien, R.Y., & Taylor, S.S. (1995) Identification of a signal for rapid export of proteins from the nucleus. Cell, 82, 463–473.
11) Fornerod, M., Ohno, M., Yoshida, M., & Mattaj, I.W. (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell, 90, 1051–1060.
12) Görlich, D. & Mattaj, I.W. (1996) Nucleocytoplasmic transport. Science, 271, 1513–1518.
13) Zhong, H., SuYang, H., Erdjument-Bromage, H., Tempst, P., & Ghosh, S. (1997) The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell, 89, 413–424.
14) Vanden Berghe, W., Dijsselbloem, N., Vermeulen, L., Ndlovu, M.N., Boone, E., & Haegeman, G. (2006) Attenuation of mitogen- and stress-activated protein kinase-1-driven nuclear factor-kappaB gene expression by soy isoflavones does not require estrogenic activity. Cancer Res., 66, 4852–4862.
15) Zhong, H., Voll, R.E., & Ghosh, S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell, 1, 661–671.
16) Oeckinghaus, A., Postler, T.S., Rao, P., Schmitt, H., Schmitt, V., Grinberg-Bleyer, Y., Kühn, L.I., Gruber, C.W., Lienhard, G.E., & Ghosh, S. (2014) κB-Ras proteins regulate both NF-κB-dependent inflammation and Ral-dependent proliferation. Cell Rep., 8, 1793–1807.
17) Beel, S., Kolloch, L., Apken, L.H., Jürgens, L., Bolle, A., Sudhof, N., Ghosh, S., Wardelmann, E., Meisterernst, M., Steinestel, K., et al. (2020) κB-Ras and Ral GTPases regulate acinar to ductal metaplasia during pancreatic adenocarcinoma development and pancreatitis. Nat. Commun., 11, 3409.
18) Postler, T.S., Wang, A., Brundu, F.G., Wang, P., Wu, Z., Butler, K.E., Grinberg-Bleyer, Y., Krishnareddy, S., Lagana, S.M., Saqi, A., et al. (2023) A pan-cancer analysis implicates human NKIRAS1 as a tumor-suppressor gene. Proc. Natl. Acad. Sci. USA, 120, e2312595120.
19) Tago, K., Ohta, S., Aoki-Ohmura, C., Funakoshi-Tago, M., Sashikawa, M., Matsui, T., Miyamoto, Y., Wada, T., Oshio, T., Komine, M., et al. (2021) K15 promoter-driven enforced expression of NKIRAS exhibits tumor suppressive activity against the development of DMBA/TPA-induced skin tumors. Sci. Rep., 11, 20658.
20) 松崎 貴(2008)毛の再生技術と創薬研究へのアプローチ.YAKUGAKU ZASSHI, 128, 11–20.
21) Bogart, B., Prutkin, L., & Ocken, P.R. (1971) The localization of phorbol ester 14C acetate in papillomas that were initiated with 7,12 DMBA and promoted with phorbol ester. An electron microscopic autoradiography study. J. Invest. Dermatol., 56, 140–146.
22) Tago, K., Funakoshi-Tago, M., Ohta, S., Kawata, H., Saitoh, H., Horie, H., Aoki-Ohmura, C., Yamauchi, J., Tanaka, A., Matsugi, J., et al. (2019) Oncogenic Ras mutant causes the hyperactivation of NF-κB via acceleration of its transcriptional activation. Mol. Oncol., 13, 2493–2510.
23) Takenaka, K., Moriguchi, T., & Nishida, E. (1998) Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science, 280, 599–602.
24) Koo, T.H., Eipper, B.A., & Donaldson, J.G. (2007) Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol., 8, 29.
25) Rasche, R., Apken, L.H., Michalke, E., Kümmel, D., & Oeckinghaus, A. (2024) κB-Ras proteins are fast-exchanging GTPases and function via nucleotide-independent binding of Ral GTPase-activating protein complexes. FEBS Lett., 598, 1769–1782.
26) Chuang, T.H., Xu, X., Quilliam, L.A., & Bokoch, G.M. (1994) SmgGDS stabilizes nucleotide-bound and -free forms of the Rac1 GTP-binding protein and stimulates GTP/GDP exchange through a substituted enzyme mechanism. Biochem. J., 303, 761–767.
27) de Ruiter, N.D., Wolthuis, R.M., van Dam, H., Burgering, B.M., & Bos, J.L. (2000) Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor-Ral pathway. Mol. Cell. Biol., 20, 8480–8488.
著者紹介Author Profile
 多胡 憲治(たご けんじ)
多胡 憲治(たご けんじ)群馬大学大学院保健学研究科生体情報検査科学 教授.博士(理学).
略歴1970年に東京都で生まれて,神奈川県で育った.93年埼玉大学理学部生化学科卒業.98年東京工業大学生命理工学研究科バイオサイエンス専攻博士後期課程修了,博士(理学)を取得.その後は,98年自治医科大学生化学講座病態生化学部門助手,2002年米国St. Jude小児研究病院Research Associate, 06年奈良先端科学技術大学院大学バイオサイエンス研究科助教,12年自治医科大学生化学講座構造生化学部門講師を経て現職.
研究テーマと抱負細胞内シグナル伝達の制御系の破綻が原因となる,がんをはじめとする様々な疾患の発症メカニズムを少しでも明らかにしたいと思い研究をしています.生化学的な実験手法を用いた泥臭いアプローチが好きです.
ウェブサイトhttps://researchmap.jp/peteneko1975