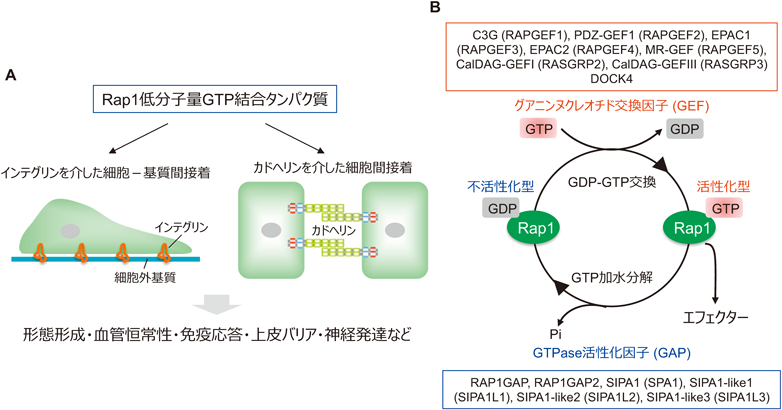
他のGタンパク質と同様に,Rap1はGDP結合不活性型からGTP結合活性型へと変換されることでエフェクター分子に結合し,下流へシグナルを伝達する2).Gタンパク質の活性は,GDPからGTPへの変換を促進するグアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)と,Gタンパク質のGTP加水分解活性を促進するGTPase活性化因子(GTPase activating protein:GAP)によって制御される(図1B).Rap1 GEFとして,触媒領域であるCdc25ホモロジードメインを有するC3G(RAPGEF1), PDZ-GEF1(RAPGEF2), EPAC1(RAPGEF3), EPAC2(RAPGEF4), MR-GEF(RAPGEF5), CalDAG-GEFI(RASGRP2), CalDAG-GEFIII(RASGRP3)が同定されている4, 10, 11).また,Cdc25ホモロジードメインを持たず異なる様式でRap1を活性化するDOCK4も存在する12).これらのRap1 GEFは,さまざまな上流シグナルによって活性化される.たとえば,C3Gはインテグリン接着によりCrkアダプタータンパク質を介して活性化され,EPAC1やEPAC2はサイクリックAMP(cAMP),CalDAG-GEF1やCalDAG-GEFIIIはカルシウムやジアシルグリセロールといったセカンドメッセンジャーによって活性化される10, 13, 14).これらの知見は,Rap1の活性が多様な上流シグナルによって厳密に制御されており,Rap1が細胞機能の制御において重要な分子であることを示唆している.一方,Rap1 GAPとしては,RAP1GAP, RAP1GAP2, SIPA1(SPA1), SIPA1-like1(SIPA1L1), SIPA1-like2(SIPA1L2), SIPA1-like3(SIPA1L3)が報告されており,それらの活性は転写調節,リン酸化などの翻訳後修飾,タンパク質間相互作用などによって制御されている4).
3. Rap1による血管バリア機能性制御メカニズム
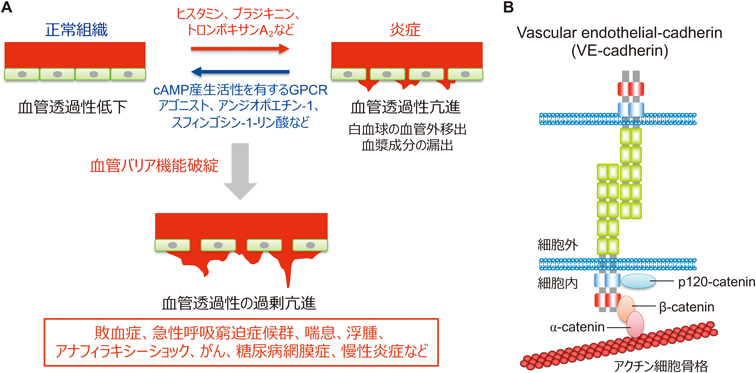
血管の内腔面では,血管内皮細胞が互いに接着することでバリア機能を有するシートを形成し,血管透過性を低い状態に維持している.末梢組織の血管では,内皮特異的な接着分子vascular endothelial(VE)-cadherin(別名Cadherin-5,CD144)が,細胞間接着を形成し,血液と血管外との物質移動を制限することで,生体恒常性を維持している15).正常組織では,VE-cadherinが強固な細胞間接着を形成し,血管透過性を低い状態に保っているが,炎症が誘導されるとVE-cadherin接着の減弱により血管透過性が亢進し,免疫細胞の血管外移出や血漿成分の漏出が誘導される(図2A)16).そのため血管内皮細胞には,血管透過性をダイナミックかつ厳密に制御するための仕組みが備わっており,その制御機構の破綻は,敗血症,急性呼吸窮迫症候群,喘息,浮腫,アナフィラキシーショック,がん,糖尿病網膜症,慢性炎症などさまざまな疾患の発症や進展と密接に関連している(図2A)17).
古典的カドヘリンファミリーに属するVE-cadherinは,1回膜貫通型タンパク質であり,細胞外ドメイン,膜貫通ドメイン,細胞内ドメインから構成される(図2B)15).細胞外ドメインは五つのカドヘリンリピートで構成され,VE-cadherinはカルシウム依存的にシス二量体を形成する.さらに,隣接する細胞のシス二量体どうしが細胞外ドメインを介してトランス結合することで細胞間接着を形成する.VE-cadherinの接着は,細胞内ドメインに結合するp120-cateninとβ-cateninによって制御される.p120-cateninはVE-cadherinに結合することで,そのエンドサイトーシスを抑制する18).一方,β-cateninはα-cateninを介してアクチン細胞骨格と相互作用し,VE-cadherin接着装置をアクチン細胞骨格に固定する(図2B)19).そのため,アクチン細胞骨格はVE-cadherin接着,および血管透過性の制御にきわめて重要な役割を果たしている.我々は,VE-cadherin接着を制御するシグナル伝達機構について研究し,Rap1の重要性を明らかにした.
1)Rap1によるVE-cadherin接着制御メカニズム
以前から,セカンドメッセンジャーであるcAMP産生活性を有するGタンパク質共役型受容体(G-protein-coupled receptor:GPCR)アゴニストが,血管透過性を抑制することが知られていた20, 21).我々は,cAMPが血管透過性を制御する機構について解析を行い,cAMPはRap1を介して,血管バリア機能を増強することを発見した22).cAMPは,下流エフェクターとして,protein kinase A(PKA)およびRap1 GEFであるEPAC1を活性化する.各エフェクター特異的cAMPアナログを用いたin vitro解析から,血管内皮細胞において,cAMPはPKAでなく,主にEPAC1を介して,VE-cadherin接着を増強し,血管透過性を低下させることを見いだし,また,EPAC1の下流因子としてRap1を同定した22, 23).すなわち,アドレノメジュリンなどのcAMP産生活性を有するGPCRアゴニストは,cAMP-EPAC1-Rap1系を介してVE-cadherin接着を増強し,血管透過性を低下させることが明らかになった.また,Rap1は,VE-cadherin接着そのものによっても活性化され,VE-cadherin接着の成熟化に寄与する24).細胞間接着によりトランス結合したVE-cadherinは,β-cateninを介してアダプタータンパク質MAGI-1を接着部位に動員する.MAGI-1は,さらにRap1 GEFの一つであるPDZ-GEF1をリクルートし,細胞間接着部位でRap1を活性化することでVE-cadherin接着の成熟を促す.
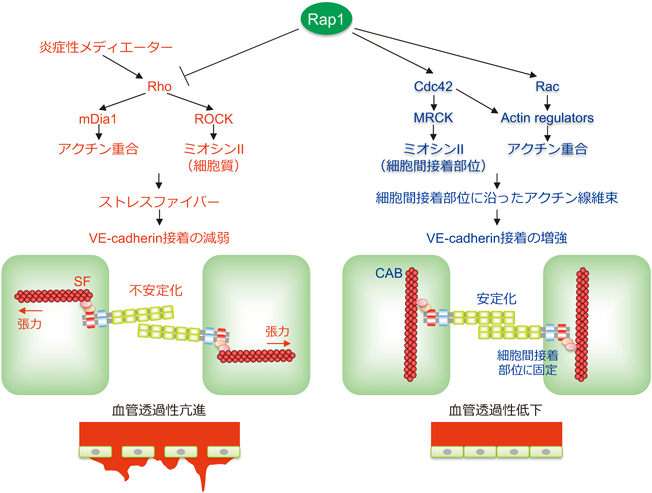
さらに,Rap1がVE-cadherin接着を増強する機構を解析した結果,Rap1はアクチン細胞骨格を劇的に再編することでVE-cadherin接着を制御することが明らかとなった(図3)5, 19, 25).ヒスタミン,ブラジキニン,トロンボキサンA2などの炎症性メディエーターは,Rho低分子量Gタンパク質を介して収縮性アクチン線維であるストレスファイバー(stress fiber:SF)の形成を促し,血管透過性を亢進させる26, 27).Rho依存的に形成されたSFは,細胞間接着部位と直交するようにVE-cadherinに結合し,張力を生じることでVE-cadherin接着を不安定化させ,血管透過性を亢進させる.一方,Rap1はRhoの活性を抑制することで,炎症性メディエーターによるSFの形成を阻害し,VE-cadherin接着の安定性を維持する25).Rap1によるRho抑制に関わるエフェクター分子として,Ras-interacting protein 1(Rasip-1)やRas-association and dilute domain-containing protein(Radil)が同定されている5, 28).Rasip-1やRadilは,Rap1依存性にRho GAPの一つArhGAP29を活性化し,Rhoを不活性化する28, 29).さらに,Rap1は,Rhoファミリーの別の低分子量Gタンパク質であるCdc42を活性化し,細胞間接着部位に沿ったアクチン線維束(circumferential actin bundle:CAB)を形成する25).このとき,Rap1はCdc42 GEFの一つFYVE, RhoGEF and PH domain containing 5(FGD5)を介して,細胞間接着部位におけるCdc42を活性化し,CAB形成を促進する.CABは,α-/β-cateninを介してVE-cadherinを細胞間接着部位に固定し,バリア機能の高い細胞間接着装置を構築することで,血管透過性を低い状態に維持する.すなわち,Rap1はRhoを抑制することでSFの形成を抑え,その一方で,Cdc42依存性にCABを形成することでVE-cadherin接着を増強し,血管バリア機能を強めている(図3).
Rap1は,アクトミオシン系の空間的な制御を介して,SF形成を阻害し,CAB形成を促進する(図3)25).Rhoは,mDia1などのアクチン重合核形成因子フォルミンを介してアクチン重合を促進するとともに,Rhoキナーゼ(ROCK)依存的に細胞質内でミオシンIIを活性化することでアクチン線維に張力を負荷し,収縮性の高いSFを形成する30).一方,Rap1はCdc42依存的にmyotonic dystrophy kinase-related CDC42-binding kinase(MRCK)を活性化し,細胞間接着部位におけるミオシンII活性を亢進することでCABの形成・維持に関与する25).CABを構成するアクチン線維の形成には,Cdc42により活性化されるアクチン重合核形成因子neuronal Wiskott-Aldrich syndrome(N-WASP)やFormin-like 3が関与すると考えられる31, 32).また,Rap1はCdc42に加えてRacも活性化することから,Racを介したアクチン重合もCAB形成に関与する可能性がある33).以上のことから,Rap1は細胞質におけるアクトミオシン系を抑えることでSF形成を阻害し,細胞間接着部位におけるアクトミオシン系を活性化することでCAB形成を誘導することが明らかとなった(図3)5, 25).
2)肺胞毛細血管のバリア機能維持におけるRap1の役割
生体においてRap1が血管透過性を制御するかを明らかにするため,タモキシフェン誘導性Cre/loxPシステムを用いて,内皮細胞におけるRap1aおよびRap1b遺伝子を欠損させた内皮特異的Rap1欠損マウスを作製し,解析を行った.同マウスはタモキシフェン投与後,数週間で死亡することがわかった.死因が血管透過性の過剰な亢進であるかを検証するため,Evans Blue血管外漏出実験を実施した.その結果,Evans Blueの漏出は肺と心臓で認められたが,脳では認められなかった.特に,肺におけるEvans Blue漏出は顕著であり,それと一致して,内皮特異的Rap1欠損マウスは肺水腫を呈していた.このことから,同マウスは肺胞毛細血管のバリア機能が破綻し,それに伴う重度の肺水腫によって死亡すると考えられた34).
Rap1が肺胞毛細血管のバリア機能を維持する機序を明らかにするため,肺動脈のホールマウント蛍光免疫染色実験を行った.その結果,野生型マウスの肺動脈ではCABが形成され,細胞間接着部位にVE-cadherinが集積していたのに対し,内皮特異的Rap1欠損マウスの肺動脈ではSFが形成され,VE-cadherinはジッパー状の局在を示していた.しかし,同マウスから肺動脈を摘出し,ex vivoでROCK阻害剤を処理すると,SFが消失し,CAB形成が部分的に回復した.さらに,肺胞毛細血管内皮細胞におけるVE-cadherinの局在を3次元蛍光免疫染色実験で解析した結果,野生型マウスではVE-cadherinが細胞間接着部位に集積し,直線状に局在していたのに対し,内皮特異的Rap1欠損マウスの肺胞ではVE-cadherinがジッパー状の局在を示し,VE-cadherin接着が破綻している様子が観察された.以上の結果から,Rap1はRhoを抑制することでSF形成を阻害し,それとともにCAB形成を促進することでVE-cadherin接着を強化し,肺胞の血管バリア機能を維持していることが明らかとなった34).肺胞毛細血管内皮細胞において,Rap1がCAB形成を促進する機序については今後の解析が必要である.
肺は,呼吸により常に外部環境にさらされているため,感染や炎症に対して脆弱である.たとえば,新型コロナウイルス感染症に罹患し重症化した患者の多くは,肺胞毛細血管のバリア機能の破綻により,非心原性肺水腫である急性呼吸窮迫症候群(acute respiratory distress syndrome:ARDS)を発症し,死に至ることもある35).我々は,Rap1を基軸とした肺胞毛細血管バリア機能維持シグナルがARDSの病態に対して保護的に作用するかを検証するため,Rap1a・Rap1bの二つのRap1遺伝子のうち,Rap1aの1アレルのみ正常な内皮特異的Rap1シグナル低下マウスを作製した.同マウスは,ストレス負荷がない状態では野生型マウスと同様に正常であったが,リポポリサッカライド(LPS)投与により,野生型マウスと比較して肺胞毛細血管の透過性が顕著に亢進した.また,EPAC1の活性化剤である007を静脈投与すると,LPS誘導性急性肺障害に伴う肺胞血管の透過性亢進が軽減された.以上の結果から,Rap1を基軸とした肺胞毛細血管バリア機能維持シグナルがARDSの病態に対して保護的に作用すること,さらにRap1シグナルがARDSの治療標的となる可能性が示唆された34).
正常組織において,cAMP-EPAC1-Rap1シグナルは血管バリア機能の維持に寄与しており,炎症性サイトカインがこのシグナルを標的とすることで血管透過性を亢進する可能性がある.以前の研究により,tumor necrosis factor-α(TNF-α)などの炎症性サイトカインは,phosphodiesterase(PDE)の発現誘導または活性化を介してcAMPを分解し,血管透過性を亢進させることが報告されている36, 37).したがって,炎症性サイトカインは,cAMPの分解を介してRap1経路を抑制し,血管バリア機能を低下させることで血管透過性を亢進している可能性がある.また,がん組織ではvascular endothelial growth factor(VEGF)が腫瘍血管の透過性を亢進させることが知られているが,この過程にもcAMP-EPAC1-Rap1シグナルの低下が関与する可能性がある.Yamauchiらは,VEGFが血管内皮細胞におけるPKA活性を低下させることで血管透過性を亢進させることを報告している38).PKAはcAMPの下流エフェクターであることから,VEGFはcAMPレベルを低下させることで血管透過性を亢進している可能性がある.したがって,腫瘍血管においてVEGFはcAMPレベルを低下させることでRap1経路を抑制し,血管透過性を亢進しているのかもしれない.これらの知見から,cAMP-EPAC1-Rap1シグナルは正常組織の血管バリア機能を維持する重要な経路であり,炎症やがんにおいてこのシグナルが抑制されることで血管透過性が亢進する可能性が示唆される.実際に,我々はcAMP-EPAC1-Rap1シグナルが正常組織の血管バリア機能維持に関与することを示唆する知見を得ている(未発表).
4. Rap1によるインテグリン接着制御メカニズム
Rap1はinside-outシグナルにより細胞内からインテグリンの接着活性を亢進し,さまざまな細胞の接着を制御する2).たとえば,血管内皮細胞ではRap1はインテグリンを活性化することで細胞–基質間接着を亢進する.リンパ球や好中球などの免疫細胞では,血管内皮細胞への接着とそれに続く血管外移出を制御するとともに,血小板の凝集や神経細胞のシナプス形成にも関与することが知られている6, 7).免疫細胞では,Rap1はRap1-interacting adaptor molecule(RIAM)に結合することで,Talinをインテグリンの細胞膜近傍に動員し,その構造を高親和型へと変換させる39).また,RapLを介してTalin非依存的にインテグリンを活性化することも報告されている40).一方,RIAMの発現は主に血球系の細胞に限定されているため,血管内皮細胞を含む血球系以外の細胞では,Rap1がTalinと直接結合し,インテグリンを活性化すると考えられている41).
1)血管内皮細胞におけるRap1依存性インテグリン接着による肺胞形態形成メカニズム
我々は,発生期および成長期におけるRap1の役割を解析するため,マウス出生直後から血管内皮細胞においてRap1aおよびRap1bを欠損させた.その結果,同マウスでは肺胞血管に顕著な異常は認められなかったものの,肺胞形成不全を呈することが確認された42).本知見は,血管内皮細胞がRap1依存的に肺胞の形態形成を制御していることを示唆している.
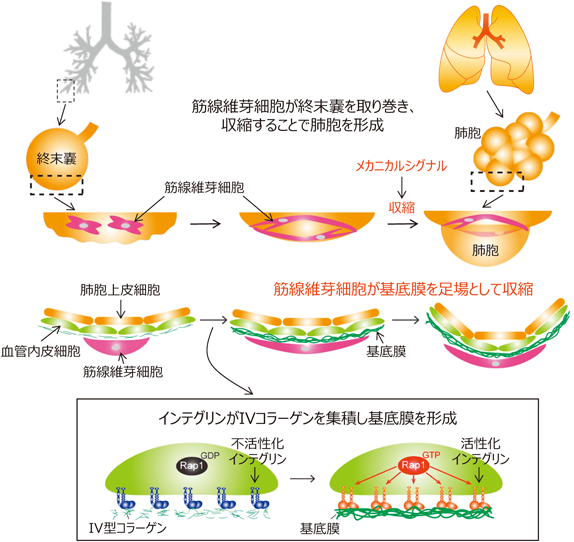
肺は,呼吸における酸素と二酸化炭素の交換を担う生命維持に不可欠な臓器であり,このガス交換が行われる場が「肺胞」である.肺胞は小さな袋状の構造をしており,その内面を覆う肺胞上皮細胞と,それを裏打ちする血管内皮細胞が密接に接着することで血液-ガス関門を形成し,肺胞内の空気と血液との間のガス交換を担う.肺胞の形成は,マウスでは生後に開始するとされており,強い収縮力を持つ肺胞筋線維芽細胞が,終末嚢と呼ばれる袋状の構造に巻きつき,収縮することで肺胞が形成されると考えられている(図4)43).これまでの研究により,肺胞毛細血管内皮細胞の発生が阻害されたマウスでは,筋線維芽細胞が存在しているにもかかわらず肺胞形成が阻害されることが報告されていた44).しかし,内皮細胞がどのように肺胞形成を制御しているのかは,これまで解明されていなかった.
血管内皮細胞がRap1依存的に肺胞の形態形成を制御する機構を明らかにするため,内皮特異的Rap1欠損マウスの肺胞筋線維芽細胞を解析した.その結果,同マウスの肺では終末嚢を取り巻く筋線維芽細胞は存在していたものの,野生型マウスと比較して,収縮に必要なアクチン線維の形成が減弱していることが示された42).筋線維芽細胞は,アクトミオシン系や核転写共役因子YAPによって構成されるメカニカルシグナルを活性化することで,アクチン線維に張力を与え,収縮する45).野生型マウスの肺胞筋線維芽細胞では,ミオシン軽鎖のリン酸化やYAPの核内移行が観察され,メカニカルシグナルの活性化が認められた.一方,内皮特異的Rap1欠損マウスの肺胞では,ミオシン軽鎖のリン酸化やYAPの核内移行が障害されており,メカニカルシグナルの活性が抑制されていることが示された42).これらの結果から,内皮特異的Rap1欠損マウスでは,筋線維芽細胞におけるメカニカルシグナルの不全により,肺胞形成が抑制されていることが明らかとなった.
そこで,血管内皮細胞のRap1が肺胞筋繊維芽細胞におけるメカニカルシグナル活性化をどのように制御するのか明らかにするため,肺胞形成期におけるこれら細胞の空間的な位置関係を3次元免疫染色法により解析した.その結果,肺胞上皮細胞が肺胞や終末嚢の内腔を形成し,その周囲を血管内皮細胞が被覆していること,さらに,外側に筋繊維芽細胞が血管内皮細胞を覆っていることがわかった42).血管の反管腔側には,基底膜が形成され,血管構造の維持に寄与していることから,筋繊維芽細胞と血管内皮細胞は基底膜を介して接していると考えられる.そこで,基底膜の主要な構成成分であるIV型コラーゲンの局在を免疫染色法により観察したところ,野生型マウスでは両細胞間にIV型コラーゲンが高度に重合した基底膜が形成され,筋線維芽細胞を密接に被覆していた.一方,内皮特異的Rap1欠損マウスでは,IV型コラーゲンの重合が弱く,基底膜の形成不全により筋繊維芽細胞との接着も阻害されていた42).基底膜は,物理的に剛直であり,細胞の足場として機能することで組織の形態形成を制御する46).また,細胞は基底膜に接着する際にその硬さを感知し,自身のメカニカルシグナルを活性化することが報告されている47).以上の結果から,血管内皮細胞は筋繊維芽細胞の足場となる基底膜を形成することで,肺胞形成を制御していると考えられる.
次に,血管内皮細胞がRap1依存性に基底膜を形成するメカニズムについて解析した.基底膜の主要な構成成分として,IV型コラーゲン,ラミニン,ニドゲン,ペルレカンが知られている.IV型コラーゲンとラミニンは自己重合する性質を持ち,重合したラミニンがインテグリンなどを介して細胞膜に結合する48).その後,IV型コラーゲンがニドゲンやペルレカンなどの因子を介してラミニンと結合することで,基底膜が形成されると考えられている.しかし,Jayadevらは,線虫の咽頭部において,Rap1がインテグリン接着を活性化し,活性化したインテグリンがIV型コラーゲンをリクルートすることで基底膜を形成することを報告した49).そこで,肺胞形成期においても,Rap1がインテグリン接着を亢進することで基底膜の形成に関与しているかを検討した.その結果,野生型マウスの肺胞形成期の血管内皮細胞では活性化インテグリンβ1が検出されたのに対し,内皮特異的Rap1欠損マウスでは,その活性が低下していた.また,野生型マウスの肺から単離した血管内皮細胞は,培養皿上で活性化インテグリンβ1陽性の焦点接着を形成したのに対し,内皮特異的Rap1欠損マウス由来の内皮細胞では,その形成が顕著に抑制されていた.さらに,ヒト臍帯静脈血管内皮細胞をマトリゲル上に播種し,血管様構造を形成させたところ,血管壁にIV型コラーゲンを成分とする基底膜様構造が形成された.一方,RAP1をノックダウンすると,この基底膜様構造の形成は消失した.しかし,インテグリンの活性化剤であるMn2+の存在下では,その形成が部分的に回復した.以上の結果から,肺胞形成期において,血管内皮細胞はRap1依存的にインテグリンβ1を活性化し,IV型コラーゲンを動員することで基底膜を形成していることが示唆された(図4)42).これを裏づけるように,内皮特異的インテグリンβ1欠損マウスでは,内皮特異的Rap1欠損マウスと同様の表現型が観察され,基底膜の形成不全により筋線維芽細胞のメカニカルシグナルが低下し,肺胞形成不全を呈することが示された42).
本研究により,血管内皮細胞においてRap1は,インテグリンの接着活性を亢進し,IV型コラーゲンを細胞膜近傍にリクルートすることで基底膜の形成を促進することが明らかとなった(図4).さらに,筋線維芽細胞は,内皮細胞が形成した基底膜を足場としてメカニカルシグナルを活性化し,収縮することで肺胞を形成することが示された(図4).これまで,血管内皮細胞はアンジオクラインファクターと呼ばれるシグナル伝達分子を産生し,臓器や組織の形成や再生を制御することが知られていた.本研究により,血管内皮細胞はアンジオクラインファクターの産生に加えて,自身の細胞機能を駆使することで,臓器や組織の形態形成を制御していることが明らかとなった.
2)造血幹細胞の発生におけるRap1の役割
ゼブラフィッシュは,臓器の発生や構造がヒトと類似した脊椎動物であり,さらに,体外受精を行い,発生が早く,胚が透明であることから,近年,生命科学・医学研究における有用なモデル動物として広く利用されている.血管系や造血系の発生プロセスの多くもゼブラフィッシュと哺乳動物の間で保存されており,これまでゼブラフィッシュを用いた研究により,多くの重要な発見がなされてきた50, 51).我々は,rap1を欠損したゼブラフィッシュの解析から,Rap1はインテグリン接着を増強することで造血幹細胞を産生する造血性内皮細胞(hemogenic endothelium)の発生を制御することを明らかにした52).
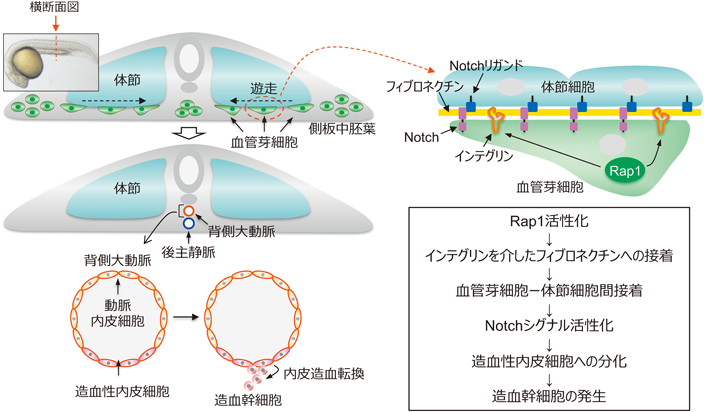
血管内皮細胞と血球細胞は,ともに中胚葉に由来し,血管芽細胞と呼ばれる共通の前駆細胞から発生する(図5).ゼブラフィッシュ胚では,両側の側板中胚葉から血管芽細胞が正中線へ移動・凝集し,受精後約20時間で頭尾軸に沿ったひも状の構造体である血管帯を形成する.その後,この血管帯は背側大動脈と後主静脈を構築し,受精後約23時間から背側大動脈の腹側に,造血幹細胞への分化能を有する特殊な内皮細胞「造血性内皮細胞」が出現する(図5)53).造血性内皮細胞は,その後,内皮造血転換(endothelial-to-hematopoietic transition:EHT)というプロセスを経て血管壁から剥離し,造血幹細胞へと分化する(図5)54).内皮造血転換は,ゼブラフィッシュにおいても保存されており,血管内皮細胞と造血幹細胞を異なった蛍光タンパク質で標識したゼブラフィッシュ胚のライブイメージングにより,背側大動脈の腹側に造血性内皮細胞が出現し,血管壁から剥離することで球状の造血幹細胞が作られることが示されている55).
血管芽細胞は,側板中胚葉から正中線へ移動する過程でNotchシグナルを受容し,造血性内皮細胞への運命を獲得することが知られている56).Notchリガンドは細胞膜上に存在するため,Notchシグナルの活性化には細胞間接着が必要である.血管芽細胞は,側板中胚葉から正中線へ遊走する際に,体節の腹側領域を通過する.この体節細胞は,NotchリガンドであるDeltaC(Dlc)およびDeltaD(Dld)を発現しており,血管芽細胞は移動の過程でこれらのリガンドを発現する体節細胞と接触することで,自身のNotch受容体を活性化し,造血性内皮細胞としての運命を獲得する(図5)57).
我々は,生体におけるRap1の機能を明らかにするため,主に血管内皮細胞で高発現するrap1bを欠損させたゼブラフィッシュを樹立した52).同フィッシュでは,顕著な血管形成異常は認められなかったものの,頭部血管に浮腫を呈する傾向がみられた.この知見から,ゼブラフィッシュにおいてもRap1が血管内皮細胞のバリア機能を制御している可能性が示唆される.さらに詳細な解析の結果,rap1b欠損ゼブラフィッシュでは,造血性内皮細胞および造血幹細胞の発生が阻害されていることが明らかとなった52).そこで,同フィッシュにおける造血幹細胞の発生異常の原因を明らかにするため,造血性内皮細胞の運命決定に重要なNotchシグナルを解析したところ,血管芽細胞が正中線で形成する血管帯において,Notch活性の低下が認められた.また,血管芽細胞においてrap1bを人為的に発現させると,rap1b欠損ゼブラフィッシュにおける造血性内皮細胞の発生異常が部分的に回復した.このことから,Rap1bは細胞自律的にNotchシグナルを調節し,造血性内皮細胞の運命決定を制御していることが示唆された.次に,rap1b欠損ゼブラフィッシュにおける血管芽細胞のNotch活性化異常の原因を明らかにするため,血管芽細胞が側板中胚葉から正中線に向かって移動する様子をライブイメージングにより観察した.その結果,rap1b欠損ゼブラフィッシュでは,血管芽細胞の遊走異常が認められ,血管芽細胞と体節細胞の細胞間接着が減弱していることが示された.以上の結果から,Rap1bは血管芽細胞とNotchリガンドを発現する体節細胞の細胞間接着を増強することで,血管芽細胞のNotchシグナルを活性化し,造血性内皮細胞への運命決定を制御していることが示唆された(図5)52).
さらに,Rap1bが血管芽細胞と体節細胞の細胞間接着を制御する機序について解析した.上述のとおりRap1はカドヘリン接着を促進するが,血管芽細胞と体節細胞の接着部位には,カドヘリン接着に必要なβ-cateninの局在が認められなかった.これらの結果から,Rap1bはカドヘリン接着を増強することで両細胞の接着を制御しているのではないことが示唆された.これまでの研究により,体節境界部にインテグリンのリガンドであるフィブロネクチンが局在することが報告されている58).そこで,Rap1bがインテグリン接着を介して,血管芽細胞と体節細胞の細胞間接着を制御しているか検討した.その結果,血管芽細胞は体節境界部に局在するフィブロネクチンに接着しながら正中線に向かって移動していることが示された52).また,ゼブラフィッシュから単離した血管芽細胞をフィブロネクチンでコートしたディッシュに播種したところ,野生型フィッシュ由来の血管芽細胞は焦点接着を形成したのに対し,rap1b欠損ゼブラフィッシュ由来の血管芽細胞では,焦点接着の形成能が低下していた52).そこで,Rap1bはインテグリン接着を活性化することで,血管芽細胞のフィブロネクチンへの接着を促進し,造血性内皮細胞の運命決定を制御しているか検証した.その結果,インテグリンβ1を欠損したゼブラフィッシュでは,rap1b欠損ゼブラフィッシュと同様に,血管芽細胞の遊走異常および造血性内皮細胞・造血幹細胞の発生不全が認められた52).また,モルフォリノオリゴヌクレオチドによるインテグリンβ1およびフィブロネクチン遺伝子のノックダウンは,血管芽細胞–体節細胞間の接着低下,血管芽細胞におけるNotchシグナル活性の低下,造血性内皮細胞・造血幹細胞の発生異常を誘導することが明らかになった52).
以上の結果から,Rap1bはインテグリン接着活性を促進することで,血管芽細胞を体節境界部のフィブロネクチンに効率的に接着させ,血管芽細胞とNotchリガンドを発現する体節細胞との接着を亢進すること,さらにこの細胞間接着の亢進が血管芽細胞におけるNotchシグナルの活性化を促し,それにより血管芽細胞が造血性内皮細胞の運命を獲得することが示された(図5)52).