低分子量GTP結合タンパク質の一つであるRhoAは,クラシカルRas homologue(Rho)ファミリーに属し(図1A),アメフラシで同定されたRho遺伝子のホモログとして哺乳類で最初に発見された分子である1).Rhoファミリーは,現在までに20種類の分子が同定されており,そのうち,12種類がクラシカル,8種類がアティピカルに分類される2).RhoAを含む低分子量GTP結合タンパク質は,一般的に,グアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)の作用によりGDPが外され,代わりに細胞内に豊富に存在するGTPが結合することで活性型となり,さらに,下流のエフェクター分子の作用を調節する3).GTPを結合した活性型の低分子量GTP結合タンパク質は,GTPase活性化タンパク質(GTPase-activating protein:GAP)により内在性のGTPaseが活性化されてGTPを加水分解しGDPとすることで不活性型となる4).
細胞レベルにおけるRhoAの主な機能は,アクチン細胞骨格の再編成である.活性型RhoAはアクチン線維を安定化して束化し,ストレスファイバーの形成を促進する.その結果,細胞内での張力が増加し,細胞の収縮能,遊走能,接着能,増殖能などが調節される5).近年,アクチン細胞骨格への作用以外のRhoAの作用についても研究が進みつつある.一方,個体レベルでのRhoAの解析は,RhoAノックアウト(knockout:KO)マウスが早期に胎生致死となってしまうこともあり,十分とはいえない6).最近では,各臓器特異的なRhoAコンディショナルノックアウト(conditional knockout:cKO)マウスを作製することで,各臓器でのRhoAの役割とその機序が徐々に明らかになってきている.
心血管系疾患による死亡は,日本では死因の第2位であり,世界ではトップの死因となっている7, 8).アジアにおける新興国などでも心血管系疾患の死亡率は増加傾向にある9).心血管系疾患におけるRhoAの関与やそのメカニズムについては不明な点が多く残されている.このような現状を踏まえて本稿では,心血管系組織におけるRhoAの作用機構について,筆者らが独自に作製したRhoA cKOマウスの解析結果を含めて紹介する.さらに,RhoAの制御分子やエフェクター分子の心血管系組織での役割についても概説し,最後に,臨床応用に向けた展開についても述べる.
心血管系組織におけるRhoAの作用について述べる前に,RhoAの分子構造について簡単に説明する.RhoAは193アミノ酸で構成される21.7 kDaのタンパク質で,細胞内に存在する.RhoAは図1Bで示すようなドメイン構造を持つ.まず,Gドメインと呼ばれるG1~G5のドメインが,GTP, GDPおよびGEFやGAPとの結合,RhoA自身の構造変化に関与する10).G1ドメインはGTP中のβ, γ位のリン酸基とMg2+イオンに結合する.G2とG3は,それぞれスイッチI,スイッチII領域内に存在し,GDPもしくはGTPが結合していることを識別して,RhoAの立体構造変化を誘導する.G4とG5ドメインがグアニン塩基と結合する.次に,スイッチI, II領域および挿入ドメインは,GEFやGAPと結合することが知られている.超可変ドメインはエフェクター分子との結合部位である.C末端モチーフ(RhoAでは,CLVLの4アミノ酸で構成)にプレニル化部位があり,RhoAの細胞膜内側への局在に重要である.スイッチI, II領域が,グアニンヌクレオチド解離阻止因子(guanine nucleotide dissociation inhibitor:GDI)のN末端領域と結合すると,スイッチI, IIの可動性が抑制され,GDPの解離が阻害される11).また,RhoGDI内の疎水性ポケットとRhoAのプレニル基が相互作用すると,RhoAは細胞膜内側への局在が阻害され細胞質に遊離する12).
1)心筋細胞レベルでの検討
心筋細胞に恒常活性型RhoAを過剰発現させた場合,やや相反する結果が同一研究グループから報告されている.2007年の報告では,アデノウイルスベクターを使って恒常活性型RhoAを心筋細胞に過剰発現させると,RhoA下流のエフェクター分子Rhoキナーゼ(Rho kinase:ROCK)を介して,Baxやカスパーゼ-9が活性化し,アポトーシスが誘導されることから,恒常活性型RhoAは心筋細胞障害的に作用すると考えられる13).一方,2008年の報告では,同様に恒常活性型RhoAを心筋細胞に過剰発現させると,FAK,ホスファチジルイノシトール3-キナーゼ,Aktのキナーゼ群が順に活性化し,心筋細胞の細胞生存が促進されるので,恒常活性型RhoAは心筋細胞保護的に作用するといえる14).別グループの研究では,薬剤により心筋細胞でのRhoAの作用を阻害すると,Aktの活性化は抑制されてカスパーゼ-3が活性化しアポトーシスが誘導されるため,RhoAは心筋保護に寄与しているとの結論になる15).また,RhoAは心筋細胞内でホスホリパーゼDと直接結合することが見いだされており,RhoAと結合できないホスホリパーゼDを心筋細胞に過剰発現させると,酸素のない虚血状態を模した環境下で細胞死がより増加したことから,RhoAはホスホリパーゼDと結合することで心筋虚血に対して保護的役割を果たしている16).
2)個体レベルでの検討
個体でのRhoAの役割についての解明は,前述のように,現在のところ十分とはいいがたいが,いくつかの報告がある.ニワトリ胚を使ったin situハイブリダイゼーション実験により,発生初期の心臓形成領域でRhoA mRNAが高発現していることが示された17).さらに,RhoAに対する低分子干渉RNAをニワトリ胚に導入しRhoAの発現を阻害すると,原始心筒の融合に障害が生じた.このことからRhoAは心臓形成期に心臓前駆細胞の遊走制御に関わっていると考えられる.
次に,哺乳類マウスの心臓におけるRhoAの役割について,心筋特異的に野生型RhoAあるいは恒常活性型RhoAを過剰発現させたトランスジェニックマウスと,心筋特異的RhoA cKOマウスを使った研究から得られた知見がある.野生型または恒常活性型ヒトRhoAを心筋に過剰発現させたマウスは出生後早期(11~16週齢)に全例死亡した18).詳細な検討は野生型RhoAを過剰発現させたマウスを使って行われているが,これらのマウスでは全身性に浮腫を来しており,心房性ナトリウムペプチドの発現が増加し,心筋組織解析でも心房,心室ともに拡大して心筋線維化が増加している所見を認め,心不全となっていた.その原因として,心房結節および房室結節の機能が低下して徐脈となり心収縮力が低下していることが見いだされた.すなわち,マウス心筋へのRhoA過剰発現は心筋障害的に作用していた.逆に,活性型RhoAの過剰発現は心筋保護的に働くとの報告もある.Tet-offシステムにより,ドキシサイクリン非投与下で心筋特異的に恒常活性型ヒトRhoAを過剰発現するマウスにおいて,心臓冠動脈前下降枝を1時間結紮した後,結紮を解除する虚血再灌流障害を与えたところ,コントロールマウスと比較して,心筋梗塞サイズは70%も減少し,心筋細胞のダメージも軽減した19).その分子メカニズムとしてRhoAによるプロテインキナーゼDの活性化が重要であることが明らかになった.以上の研究結果は同一研究グループからのものであるが,恒常活性型RhoAの過剰発現が心筋において保護的でもあり,障害的でもある理由についてははっきりしないものの,心筋が受ける環境や状況によってRhoAの作用やシグナル伝達機構が異なるためであるかもしれない.
これに対し,心筋特異的にRhoAの発現を欠損させたRhoA cKOマウスは,筆者らの検討でもまた別グループの検討でも異常なく出生し,少なくとも若年期まではコントロールマウスと同様に成長し,心機能についても異常は認められなかった20, 21).RhoA cKOマウスに弓部大動脈縮窄(transverse aortic constriction:TAC)を施して大動脈内の血液を流れにくくし,心臓に過剰な圧負荷をかけた場合,TAC後の早期(2週間後)に誘導される心肥大の程度について,コントロールマウスとRhoA cKOマウスで差異はみられなかったが,TACから8週間後のRhoA cKOマウスでは心室が拡大し,心室壁が菲薄化して拡張型心筋症様の所見を認めた20).Rho cKOマウスの心筋では,心筋細胞内のCa2+シグナルが異常になるとともに,MAPキナーゼやAktの活性化が低下することで細胞生存機能も抑制されていた.すなわち,RhoAが複数の細胞内シグナル伝達機構を制御して心筋保護的に作用していることが見いだされた.
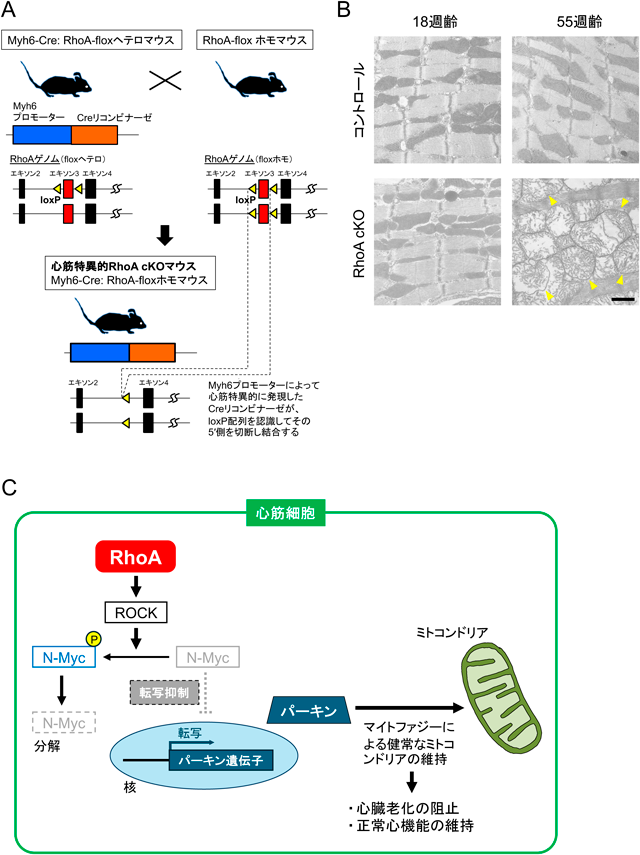
筆者らは,心筋特異的RhoA cKOマウスを独自に作製し(図2A),心筋RhoAの加齢における機能について検討した.上述のように,若年期まではRhoA cKOマウスとコントロールマウスで差異はみられなかったが,RhoA cKOマウスは30週齢ごろより死亡する個体が現れ,60週齢までに全例死亡した21).心臓超音波検査で心機能を調べると,心機能指標の一つである左室駆出率が20週齢ごろより徐々に低下し,50週齢ごろには左室駆出率は20%台となり,正常の左室駆出率(60~70%)の約1/3にまで低下した.RhoA cKOマウス心筋組織では,老化関連β-ガラクトシダーゼなどの老化マーカーの発現が有意に増加していた.電子顕微鏡による心筋細胞内の観察で,RhoA cKOマウスのミトコンドリアのほとんどが,加齢に伴って高度に損傷され構造破綻していることがわかった(図2B).すなわち,RhoAは心筋老化を制御することで心筋保護的に作用していた.その制御機構として,RhoAはパーキンを介して損傷したミトコンドリアを処理するマイトファジーを調節し,心筋の構造や機能維持に関与していることが明らかになった.RhoAの発現が欠損するとパーキンの発現が低下して,マイトファジーの機構が作動しなくなるため,損傷した異常ミトコンドリアが排除されずにそのまま蓄積し心機能低下が生じた.パーキンの発現はN-Mycにより抑制されているが,RhoAによりROCKを介してN-Mycがリン酸化されるとN-Mycはプロテアソームで分解されて減少するので,その抑制が外れてパーキンの発現が増加する(図2C).しかし,RhoAの発現が欠損していると,N-Mycはリン酸化されずに細胞内で発現が維持され,その結果,パーキンの発現は抑制され低下したままとなっていた.ところで,パーキン遺伝子の変異は家族性パーキンソン病と関連するが22),パーキンソン病では心不全のリスクが上昇するとの報告もある23).次に,RhoA cKOマウス心筋に,アデノ随伴ウイルス(adeno-associated virus:AAV)ベクターを用いてパーキンを発現させたところ,50週齢のRhoA cKOマウスでも左室駆出率は維持されて心機能の低下を阻止することができ,寿命短縮の回復につながった.さらに,原因遺伝子が特定されていない特発性拡張型心筋症により重度の心不全となり,補助人工心臓装置を装着せざるをえなくなった患者の心筋でもRhoAとパーキンの発現が両者とも有意に低下していた.以上,筆者らの研究を含めた複数の研究結果から,心筋でのRhoAの発現欠損は心機能低下につながることから,個体での心機能維持にRhoAの存在は必須であると考えられる.
1)血管内皮細胞におけるRhoAの作用機構
血管を構成する重要な細胞として内皮細胞と平滑筋細胞がある.まず,血管内皮細胞におけるRhoAの作用機構について述べる.血管内皮細胞におけるRhoAは,血管透過性に関与しているとの報告が多い.炎症を促進する脂質リゾホスファチジルコリンによる血管内皮の透過性亢進に,RhoAの過剰活性化と,その結果生じる血管内皮細胞のバリア機能障害が強く関与することが明らかになっている24).ボツリヌスC3毒素でRhoAの作用を阻害すると,リゾホスファチジルコリンによる血管内皮の透過性亢進は抑制された.また,RhoAはプロテインキナーゼCαの下流で機能していることが示された.同様に,トロンビン刺激によってもプロテインキナーゼCα活性化を介してRhoAが活性化し,それにより血管内皮細胞内のアクチン細胞骨格が再編成され,血管内皮の透過性が高まる結果となった25).重度の外傷性傷害を受けショック状態となった患者血清を,培養血管内皮細胞の培養液中に投与すると,RhoAは過剰活性化し,血管内細胞間の透過性は亢進した26).血管内皮の透過性亢進はショック患者の状態をより悪化させるため,外傷によるショック状態の患者の血清中に含まれるRhoA活性化因子を除去したり,内皮細胞でのRhoA活性化を抑制したりすることで,ショックの病態を改善し救命率向上に寄与できる可能性がある.
RhoAは血管内皮増殖因子(vascular endothelial growth factor:VEGF)による血管新生においても重要な役割を果たしているとの報告が多い.RhoA阻害薬を投与したりドミナントネガティブRhoA変異体を過剰発現させたりするin vitroやex vivoの実験系で,RhoAおよびその下流分子ROCKが,VEGF刺激による血管内皮細胞の遊走,生存,管腔形成を促進していることが示された27, 28).しかし逆に,RhoAの活性化が血管内皮細胞の遊走や生存を抑制し,血管新生に関わる管腔形成などを阻害しているとの最近の報告もあり29),現在でもRhoAの血管内皮細胞における作用や血管新生における意義については議論の余地が残されている.
内皮細胞でRhoAの発現を欠損させたRhoA cKOマウスの解析もすでに行われている.胎生期から内皮細胞でRhoAの発現を欠損させても,RhoA cKOマウスの血管形成に異常を認めなかった30).ただ,RhoA cKOマウスの出生数は有意に減少していた.さらに,薬剤誘導性に内皮細胞でRhoAの発現を欠損させたRhoA cKOマウスでは,網膜の血管新生に軽度の異常が生じた.このように,内皮細胞RhoA cKOマウスの表現型はまったく正常ということではないので,さらに詳細な解析が必要である.筆者らはこの観点から独自に内皮細胞RhoA cKOマウスを作製して解析しており,途中段階であるが,既報の結果と異なる点があるため,今後も個体レベルでの血管内皮細胞におけるRhoAの役割とその分子メカニズムについて検討を続ける予定である.また,ヒトにおいて約200人に1人の頻度でみられる脳海綿状血管腫で,特に,CCM2遺伝子に異常のある家族性脳海綿状血管腫についてマウスで解析した研究により,RhoAの活性化がこの疾患の病態に寄与していることが見いだされた31).薬剤でRhoA活性化を抑制することにより,脳海綿状血管腫による異常な血管透過性を改善できることも示された.
2)血管平滑筋細胞におけるRhoAの作用機構
RhoAの血管平滑筋細胞における機能については,その細胞収縮を制御する分子機構を中心に,これまでに多くの研究がなされ豊富な知見が蓄積されている32–35).血管平滑筋細胞を脱分極させると,RhoAが活性化して細胞収縮が生じる36).前述のように,血管内皮細胞ではプロテインキナーゼCαの活性化がRhoAの活性化を促進するが25),血管平滑筋細胞では逆に活性化したプロテインキナーゼCαはRhoAの活性化を抑制していた.また,血管平滑筋細胞の収縮を促進するセロトニンを短時間(1時間程度)血管平滑筋に作用させると細胞内で活性化したRhoAが増加するものの,長時間(48~72時間)作用させるとRhoAはプロテアソームで分解を受けて発現量が減少し,その結果,動脈の収縮性も抑制されることになった37).さらに,肺動脈の中膜を構成する血管平滑筋細胞では,その存在部位の違いにより低酸素に対するRhoA活性化度合いが異なるようである38).具体的には,胎仔または生後3日目の仔ブタの肺動脈中膜の内側から単離した血管平滑細胞では,8時間の5%低酸素状態により,活性化したRhoAが増加した.しかし,同条件でも,肺動脈中膜の外側から単離した血管平滑筋細胞でのRhoA活性化はみられなかった.一方,生後14日目の血管平滑筋細胞では,肺動脈中膜の内側あるいは外側からの単離にかかわらず,RhoAの活性化はみられず,逆に,外側から単離した血管平滑筋細胞ではRhoAの活性化は抑制される傾向にあった.その他,血管平滑筋の分化にもRhoAが関与しているとの報告がある.恒常活性型RhoAを血管平滑筋細胞に導入すると,血管平滑筋細胞の分化を調節するSM22やα-SMAのプロモーター活性が上昇したが,この活性上昇はRhoAによるアクチンの重合に依存していた39).
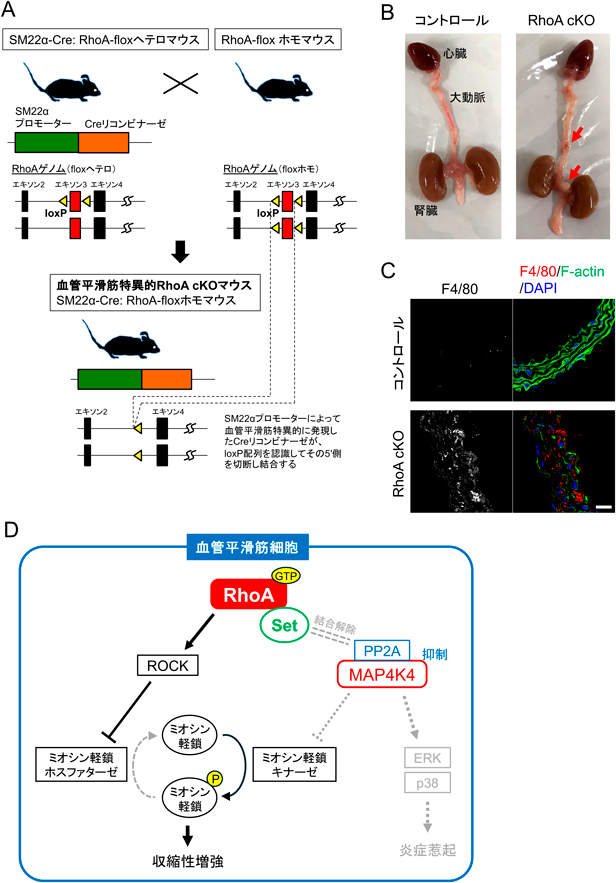
個体の血管平滑筋でのRhoAの役割を明らかにするため,筆者らは血管平滑筋特異的RhoA cKOマウスを作製して解析を行った(図3A).このRhoA cKOマウスは異常なく出生し成長した.しかし,アンジオテンシンIIとリシルオキシダーゼ阻害薬の薬剤刺激により,コントロールマウスと比較して,腹部大動脈瘤の発生率が有意に増加し(図3B),一部,その破裂で突然死する個体も現れた40).RhoA cKOマウスの大動脈では,平滑筋の強度や収縮に関わるタンパク質の発現が減少しており,大動脈の収縮性が減弱していた.さらに,大動脈での炎症性サイトカインの増加や炎症細胞の浸潤(図3C),細胞外マトリックス分解酵素の発現亢進を認めた.これらのことから,RhoA cKOマウスの大動脈は高血圧による圧負荷に対して脆弱になっていたと考えられた.その原因について検討した過程で,血管平滑筋細胞内でRhoAに結合するエフェクター分子Setを新規に同定した.SetはホスファターゼPP2Aと結合してPP2Aの作用を阻害することで,MAP4K4分子の脱リン酸化(不活性化)を抑制し,結果的にMAP4K4の活性化が維持される.しかし,RhoAが存在すると,SetはPP2AよりもGTP結合型の活性化RhoAとより強く結合するため,Setから遊離したPP2AはMAP4K4と結合してMAP4K4を不活性化する.その結果,MAP4K4によるミオシン軽鎖キナーゼの抑制が解除されて平滑筋細胞の収縮性は増強し,また,MAP4K4下流のERKやp38の活性化は抑制されるため大動脈組織での炎症は減弱する(図3D).これらの作用により,腹部大動脈瘤の形成は抑制されることになる.一方,RhoA cKOマウスではこれらの機序が破綻し,腹部大動脈瘤の発生増加につながっていると想定された.実際,RhoA cKOマウスにMAP4K4阻害薬DMX-5804を投与すると,大動脈への炎症細胞浸潤は抑制され,腹部大動脈瘤の発症も抑制された.ヒト腹部大動脈瘤の手術時切除サンプルを使った解析でも,正常部位と比較して,大動脈瘤部位でRhoAの発現が有意に低下していることが示された.また,マウスにアンジオテンシンIIを投与して血管平滑筋でRhoAが過剰活性化した状態にすると,血管のリモデリングにより大動脈の線維化などが増加したが,このマウスにドコサヘキサエン酸代謝物レゾルビンD1を投与してRhoA過剰活性化を抑制すると,大動脈の線維化を改善することができた41).以上の結果より,RhoAの適切な発現と活性化が,血管平滑筋細胞の健常性と大動脈など血管の構造・機能維持に重要であると考えられる.
5. RhoA活性化制御分子の心血管系組織における役割と分子機構
RhoAの活性化,すなわち,GTP結合型への促進に関わるのはGEFであり,RhoAの不活性化,すなわち,GDP結合型を促進・維持するのはGAPとGDIである.GEF, GAP, GDIはそれぞれ主としてRhoAの活性化,不活性化を介して心血管系組織を含めたさまざまな組織に作用するが,一部,それぞれ独自に作用する場合もある.本節では,GEF, GAP, GDIに属する分子群のうち,心血管系組織への作用が見いだされている分子のいくつかについて述べる.
1)GEF
RhoGEFは一般的に約200アミノ酸で構成されるDblホモロジーと呼ばれるGEF活性を有するドメインを持ち,このドメインがRhoAのスイッチI, II領域と結合することでRhoAの活性化に寄与する3).
心肥大に伴い心筋細胞でPDZ-RhoGEFの発現が増加するが,心筋特異的なPDZ-RhoGEF cKOマウスでは,圧負荷による心肥大や心筋線維化が抑制され,心機能低下も抑制できることが最近明らかになった42).そのメカニズムとして,PDZ-RhoGEFはGタンパク質Gα13と結合してGα13を介したRhoAの活性化と心肥大シグナルに関与することが示された.また,ゼブラフィッシュで発現するRhoGEFのEct2について,Ect2発現欠損個体の心臓では多核の心筋細胞が増え,同様に,Ect2のドミナントネガティブ変異体を発現させたトランスジェニック個体でも多核の心筋細胞が増えて,心筋細胞が過剰に肥大した43).ゼブラフィッシュでは多核の心筋細胞の割合が増加すると,心筋細胞の増殖能が抑制され心筋再生能が低下した.ただ,Ect2によるRhoA活性化がこの現象にどの程度関与しているかは不明である.
血管内皮細胞において,p115RhoGEFはRhoAの活性化を介して,リポ多糖や腫瘍壊死因子-αの作用による内皮細胞のバリア機能障害・破綻を促進する作用がある.血管内皮細胞でp115RhoGEFをノックダウンすると,アクチンストレスファイバーの形成は抑制され,内皮透過性は低下してバリア機能が回復することが見いだされた44, 45).血管内皮細胞における別のRhoGEFであるLARGは血管内皮細胞では,接着分子ICAM-1の下流で働き,RhoAの活性化を介して白血球の内皮細胞下への遊走を促進する46).また,Net1は内皮細胞の増殖や遊走,管腔形成を促進して,腫瘍の血管新生に関わることが示されている47).
血管平滑筋細胞でもいくつかのRhoGEFの機能が明らかになっている.動脈硬化を引き起こすApoEノックアウトマウスにヘテロにSmgGDSをノックアウトすると,アンジオテンシンII刺激により胸部大動脈瘤が形成され,多くの個体がその破裂により突然死した48).このダブルノックアウトマウスの大動脈平滑筋細胞では,フィブリリン-1の発現が減少し,IL-2やIL-6などの炎症性サイトカインの発現が増加して,大動脈壁が脆弱になっていたと考えられた.また,血管平滑筋に高発現するVsm-RhoGEF(別名Ephexin5)は,エフリン-A刺激によりEphA4受容体の下流で活性化し,RhoAを介して平滑筋細胞の収縮性を高めることが見いだされている49).
2)GAP
哺乳類では,現在までに約80個のRhoGAPが同定されている50).RhoGAPはRhoAのスイッチ領域に結合し,そのGTPaseを活性化させることでGTPの加水分解を促進して,RhoAを不活性化する.ここでは三つのRhoGAPについて紹介する.一つ目のp73RhoGAPは血管,特に,血管内皮細胞あるいは血管平滑筋細胞に多く発現し,血管新生を促進する51).p73RhoGAPを血管内皮細胞でノックダウンすると活性化型RhoAは増加し,内皮細胞の遊走,増殖および管腔形成能は抑制された.このGAPに対するアンチセンスオリゴを導入した内皮細胞をゲル状にしてマウス皮下に移植した場合,内皮細胞による新生血管形成が抑制された.次に,血管内皮細胞に発現するp190RhoGAPはリン酸化されることで活性化し,RhoAは不活性化される.その結果,内皮細胞の遊走は抑制され,血管内皮の透過性が調節される52, 53).血管平滑筋細胞ではアンジオテンシンIIに対するRhoAの活性化にp190RhoGAPが関与している54).アンジオテンシンII刺激に対してp190RhoGAPは脱リン酸化酵素SHP-2により脱リン酸化され不活性化されることで,逆に,活性型RhoAが増加した.三つ目のARHGAP26(別名Graf)は動脈管での作用が報告されている.新生児において動脈管が開存している場合,動脈管でのARHGAP26の発現が有意に低下していたことから55, 56),動脈管の開存・閉鎖におけるARHGAP26の重要性が示唆された.動脈管平滑筋細胞でARHGAP26の発現をノックダウンすると,活性型RhoAは増加し,動脈管を閉鎖するのに重要な平滑筋細胞の遊走や増殖が抑制された56).
3)GDI
現在までに多くのRhoGEF, RhoGAPが同定されているのに比較して,RhoGDIは哺乳類では,RhoGDI-1,-2,-3の3分子のみが同定されている.そのうちのRhoGDI-1については,肺微小血管内皮細胞でRhoAの活性化を抑制し,血管透過性を調節しているとの報告がある57).RhoGDI-1ノックアウトマウスを用いた解析で,RhoAが活性化して内皮細胞間隙が拡大し,肺微小血管の透過性が亢進することが見いだされた.この知見は,重度の炎症性肺疾患や呼吸窮迫症候群でみられる肺微小血管から間質への血漿漏出による呼吸困難を改善するのに役立つ可能性がある.
6. RhoAエフェクター分子の心血管系組織における役割と分子機構
RhoA下流のエフェクター分子がいくつか同定されているが,本節ではROCKとmDiaの二つに絞り,これらの分子の心血管系組織における作用について述べる.
1)ROCK
ウシ脳やヒト血小板サンプルから同定された約160 kDaのセリン/トレオニンキナーゼである58, 59).ROCK1とROCK2のアイソフォームが存在し,マウスでは,ROCK1は全組織にユビキタスに発現しているが,ROCK2は脳や骨格筋に多く発現する60).
ROCK1のヘテロKOマウスやホモKOマウスでは,薬剤またはTACによる心臓への過剰な圧負荷により心筋線維化が抑制され,さらに,心筋細胞のアポトーシスも抑制されたことから61–63),ROCK1の発現は心臓にとって好ましくないのかもしれない.しかし別の論文では,心筋特異的ROCK1 cKOマウスにTACにより心臓へ過剰圧負荷をかけると,心筋線維化は増大するとともに,心機能が低下して心不全になったと示されており64),ROCK1は心保護的に作用しているといえる.このように,ROCK1の心臓での役割についてははっきりしない.一方,心筋特異的なROCK2 cKOマウスでは,ROCK1 cKOマウスとは逆に,TACによる心臓への過剰圧負荷でも心筋線維化は増大せず,心機能低下も認めなかった64, 65).また,TAC後の心臓での酸化ストレスも,ROCK1 cKOマウスでは上昇するが,ROCK2 cKOマウスでは低下した.これらの結果から,心臓においてROCK1とROCK2は反対の作用を発揮している場合があると考えられる.
血管平滑筋特異的なROCK1またはROCK2 cKOマウスでは,低酸素状態で肺高血圧が生じにくくなり,逆に,ROCK2過剰発現マウスでは肺高血圧が生じやすくなった66, 67).血管平滑筋細胞において,ROCK2を介したERKの活性化やその結果促進される細胞増殖能および細胞遊走能が,ROCK2 cKOマウスでは抑制されるために肺高血圧になりにくくなるというメカニズムが示された67).肺高血圧の患者の肺動脈でROCK2の発現が上昇していることも合わせて示された.
ROCK1とROCK2のダブルKOマウスの解析も行われている.ROCK1−/−ROCK2−/−マウスは胎生早期(胎生9.5日より前)に全例死亡した68).次に,ROCK1+/−ROCK2−/−マウスまたはROCK1−/−ROCK2+/−マウスは胎生9.5~12.5日の間に死亡したが,その原因として卵黄嚢の血管形成不全が示唆された.最近,薬剤誘導により成体マウスでROCK1とROCK2をダブルKOした場合の解析結果が報告された69).これらのマウスでは肺血管内皮細胞でのVE-カドヘリン/β-カテニンの発現が低下し,内皮細胞間の細胞接着が減弱して,肺血管の透過性が亢進するため,肺胞壁が肥厚し肺機能が阻害されていることがわかった.なお,心筋特異的にROCK1とROCK2をダブルKOしたマウスでは,心筋でのオートファジーが促進し,加齢による心筋線維化が抑制される結果となった70).
2)mDia
フォルミンファミリーに属するタンパク質で,ショウジョウバエDiaphanous分子の哺乳類ホモログとしてとして見いだされ,アクチン分子の重合を促進してアクチン線維の伸長に関わっていることがよく知られている71).mDia1(別名DIAPH1),mDia2, mDia3の三つのアイソフォームが存在する.
血管内皮細胞ではmDiaは,活性化したRhoAとともに,サイクリン依存性キナーゼ阻害分子p27kip1の分解を増やすことで,細胞周期G1期からS期への移行を促進していた72).血管平滑筋細胞においてmDia1とmDia2は高発現しており,RhoAやROCKの下流で両mDia分子は平滑筋細胞に特異的な分子であるSM22, α-SMAの発現を促進し,血管平滑筋細胞の分化を制御していることが見いだされた73, 74).
次に,個体レベルの知見として,mDia1 KOマウスでは,TACによる心臓への過剰圧負荷に対して脆弱であった.具体的には,このcKOマウスでは心機能が低下し,心筋細胞内のミトコンドリアの形態異常を認め,コントロールマウスと比較して死亡率が上昇した75).すなわち,心臓へ過剰な圧負荷がかかっている状態ではmDia1は心保護的に働いていると考えられる.一方,心臓に虚血再灌流障害を与えた場合,野生型マウスの心筋でmDia1の発現は増加するとともに,mDia1 KOマウスでは心筋梗塞サイズは減少し,心機能低下もみられなかった76).この結果から,過剰圧負荷状態の場合とは逆に,虚血再灌流障害においてmDia1は増悪因子として作用することが示唆された.なお,mDia2 KOマウスは赤芽球の細胞質分裂が障害され重度の貧血により胎生12.5日目までに致死となったが77),mDia3 KOマウスには明らかな異常はみられなかった78).
7. RhoAシグナル伝達系の調節による心血管疾患への治療応用
RhoAシグナル伝達系と心血管疾患治療との関連では,ROCK阻害薬ファスジルがくも膜下出血後の脳血管れん縮に対する治療薬として使用されていることがあげられる.この脳血管れん縮のメカニズムについては解明されていない点も残されているが,内皮細胞での一酸化窒素の産生減少がメカニズムの一つと考えられている79).くも膜下出血により低酸素状態が出現するが,そのような状態でRhoAやROCKは内皮型一酸化酸素合成酵素を抑制して,一酸化窒素産生を阻害するとの報告がある80).次に,肺動脈性肺高血圧についても,ROCK阻害により症状が改善することが動物実験レベルで見いだされている81, 82).肺高血圧に対するROCK阻害薬の臨床応用について,いくつかの臨床治験も行われてきたがまだ治療薬として承認されたものはなく83),今後の展開が期待される.
自験例であるが,前述のように,心筋細胞や血管平滑筋細胞でRhoAの発現を欠損させたcKOマウスでは,それぞれ,拡張型心筋症,腹部大動脈瘤に相当する疾患発症の頻度が上昇した21, 40).拡張型心筋症や腹部大動脈瘤患者から得たヒトサンプルにおいても,RhoAの発現は減少していた.このことから,これらの疾患の予防や治療におけるRhoA補充遺伝子治療の有用性が想定される.遺伝子治療法として最近注目を集めているのが,AAVベクターを用いて目的遺伝子を標的とする臓器にできるだけ選択的に導入し,その遺伝子産物を発現させるものである84).AAVには血清型(セロタイプ)があり,それぞれの血清型によりAAVが感染しやすい臓器が異なる.日本では最近,AAV9ベクターにSMN1遺伝子を導入したオナセムノゲン アベパルボベク(商品名ゾルゲンスマ;2020年承認)が脊髄性筋萎縮症に対する治療薬として,また,AAV2ベクターにRPE65遺伝子を導入したボレチゲン ネパルボベク(商品名ルクスターナ;2023年承認)が遺伝性網膜ジストロフィーの治療薬として承認された.AAV遺伝子治療の問題点として,治療費がきわめて高価(数千万円から億単位)であることはさておき,すでに抗AAV抗体を有している患者への投与は困難であること,また,一度投与すると抗AAV抗体ができてしまうため単回投与しかできないことや,静注投与後の肝毒性などがある.これらの問題を克服のため,より免疫原性の低いAAVベクターの開発などが進められている.
本稿ではRhoAとその関連分子に焦点を当て,特に,生体での心血管系組織におけるその生理学的・病理学的役割と分子作用機構について紹介した.RhoAを含むRhoファミリー低分子量GTP結合タンパク質の研究はすでに数多くなされており,これまでに豊富な知見が得られている.それでも,RhoAシグナル系の新たな制御機構が最近次々と明らかになっており,今後もこのシグナル系に着目した研究は発展し続けていくと思われる.研究成果の解釈や活用にはさまざまなものがあるが,基礎研究の意義の一つとして疾患の病態メカニズム解明に貢献できることがあげられる.そのことは疾患に対する新たな予防法・治療法の開発につながるものである.