細胞内には,タンパク質や脂質などの物質を適切な量,適切な場所へ配送するための膜小胞を介した物流システムが存在し,細胞の機能や恒常性の維持に不可欠な役割を果たしている.神経細胞において小胞輸送系が果たす役割は,シナプス前終末における神経伝達物質の放出,神経伝達物質受容体の調節によるシナプス可塑性,神経突起におけるシグナル分子やオルガネラの極性輸送,さらには細胞内の物質の品質管理など多岐にわたる.さらに小胞輸送プロセスの破綻がアルツハイマー病やパーキンソン病などの神経変性疾患の病態機序として指摘され近年注目されている.
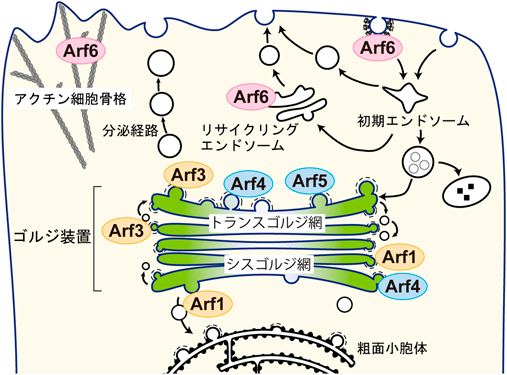
ADPリボシル化因子(Arf)は,細胞内の小胞輸送やアクチン細胞骨格の再編成を制御する約20 kDaの低分子量Gタンパク質である.哺乳類では6種類のArfタンパク質が存在し(ヒトではArf2は偽遺伝子化し存在しない),アミノ酸配列の相同性からクラスI(Arf1, Arf2, Arf3),クラスII(Arf4, Arf5),クラスIII(Arf6)の三つのサブグループに分類される1).クラスIおよびIIのArfは主にゴルジ体や小胞体に局在し,これらのオルガネラ間での分泌経路を制御している(図1).一方で,唯一クラスIIIに属するArf6は細胞膜やエンドソームに局在し,エンドサイトーシスやリサイクリング経路を含めたエンドソームと細胞膜の間の小胞輸送経路の制御を担っている.またArf6は,アクチン細胞骨格の再編成を介した細胞膜ダイナミクスにも関与する2).Arfはほかの低分子量Gタンパク質と同様に分子スイッチとして機能し,活性化型であるグアノシン三リン酸(GTP)結合型と不活性化型であるグアノシン二リン酸(GDP)結合型の二つの間を可逆的に循環する.GTPが結合したArf6は構造変化を起こし,下流のエフェクター分子に結合することでシグナルを伝達する(Arfのエフェクター分子についてはほかの総説を参照されたい1)).Arfの活性状態は2種の活性制御因子により厳密に制御されており,GDPからGTPへの変換はグアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)が担い,ArfのもつGTPase活性を促進させることでGTPを加水分解させてGDPに変換するのはGTPase活性化タンパク質(GTPase-activating protein:GAP)の役割である.Arfに対するGEFはSec7ドメインを持つことを特徴とし,哺乳類では6ファミリー15分子が同定されている.また,Arfを不活性化するGAPには共通の構造としてArf-GAPドメインを持つ10ファミリー31分子が存在する.これらの活性化制御因子はそれぞれ異なる分子構造や基質特異性,活性制御機構,組織・細胞内局在を示す1, 3).
Arf6は実にさまざまな神経機能に関与しており,神経上皮の形成や維持4),神経細胞の移動5, 6),軸索および樹状突起の形成7, 8),シナプス前終末でのシナプス小胞のリサイクル9),スパイン形態形成10–12),シナプス可塑性13)などの制御を担っている.近年の研究から,Arf6が担う多彩な神経機能とArf6を活性化するGEFの分子多様性との関連が明らかになってきている.本稿では,Arf6の活性化を制御するGEFファミリーについてそれぞれの特徴を述べた後,Arf6とそのGEFが果たす神経機能への役割について,我々の研究成果を含めた最近の知見を中心に紹介する.
2. Arf6-GEFの分子多様性と神経系での発現分布
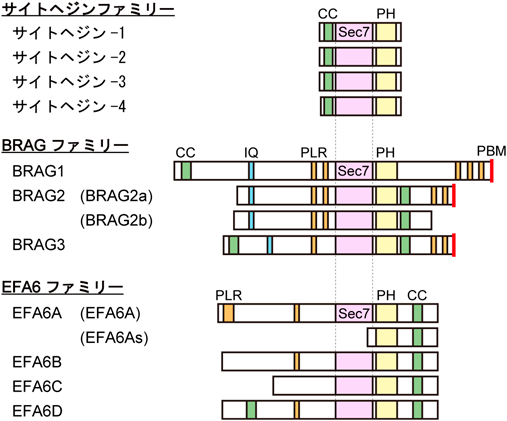
Arf6を活性化するGEFは,共通のドメイン構造としてGEF活性に必須の200アミノ酸程度からなるSec7ドメインを持つ6種のSec7タンパク質ファミリーのうち,サイトへジンとBRAG, EFA6の三つのファミリーに属する14, 15)(図2).
1)サイトへジンファミリー
サイトへジンファミリーは,約47 kDaのArf-GEFの中では比較的低分子量の分子であり,その分子構造としてN末端にコイルドコイル(CC)ドメイン,中央にSec7ドメイン,C末端にプレクストリン相同(PH)ドメインを持つ.サイトへジンファミリーは哺乳類ではサイトへジン-1およびサイトへジン-2(ARNO),サイトへジン-3(ARNO3またはGRP1),サイトへジン-4の四つの分子が同定されている16).サイトへジンファミリー分子のArfに対する基質選択性に関して議論の余地が残るが,in vitroの解析からはサイトへジン-1および2, 3がArf6に対するGEF活性を有し,in vivoにおいてもサイトへジン-1と2がArf6のGEFとして機能することを支持する知見が蓄積している17–19).
in situハイブリダイゼーション法による遺伝子発現解析から,サイトへジン-4を除いたほかのサイトへジンが多くの神経領域において複数のサイトへジンmRNAを同時に発現していることが示されている20).たとえば海馬錐体細胞や歯状回顆粒細胞はサイトへジン-1から3のいずれも,また小脳プルキンエ細胞はサイトへジン-1と3を発現している.サイトへジンファミリーのうち,タンパク質レベルでの詳細な発現局在解析がなされているのはサイトへジン-2のみである.著者らは,サイトへジン-2が海馬CA1領域の錐体細胞の細胞体や樹状突起において細胞膜やさまざまな輸送小胞(初期エンドソーム,リサイクリングエンドソーム,後期エンドソーム,リソソーム)に局在することを免疫組織化学的解析により明らかにした21, 22).さらに,サイトへジン-2は興奮性シナプスのシナプス後肥厚(postsynaptic density:PSD)周縁部にも集積することを免疫電子顕微鏡解析で見いだした21).このサイトジン-2のペリシナプスでの局在は,足場タンパク質であるタマリンを介してI型代謝型グルタミン酸受容体(mGluR1および5)と複合体を形成するという既報を支持しており23),mGluR1/5シグナルにおけるサイトへジン-2を介したArf6の機能関与が示唆される.
2)BRAGファミリー
BRAG(Brefeldin A–resistant Arf-GEF)またはIQSEC(IQ motif and SEC7 domain-containing protein)ファミリーは,哺乳類ではBRAG1/IQSEC2, BRAG2/IQSEC1, BRAG3/IQSEC3の三つの分子からなる.BRAGファミリーの構造的特徴は,N末端領域にカルモジュリン結合配列であるIQモチーフと中央にSec7ドメインを有することである24).これらのドメインに加えて,BRAG1–3はそれぞれPDZ(PSD-95/Discs large/zonula occludens-1)ドメイン結合モチーフをC末端部位に持ち,これが細胞内局在に大きく影響する.興奮性シナプスのPSDに豊富に局在するPSD-95の相互作用分子のプロテオミクス解析から,BRAG1とBRAG2が見いだされており25),BRAG1とBRAG2はPDZドメイン結合モチーフを介してPSD-95に結合することで興奮性シナプス後膜に選択的に局在することが示されている26–28).ただし,BRAG2には少なくとも二つのスプライスアイソフォームがあり(図2),BRAG2aはPDZドメイン結合モチーフを有するため興奮性シナプス後膜に選択的に局在するが,このモチーフを持たないBRAG2bは初期エンドソームなどの細胞内小胞に主に局在する26).著者らは最近,先天性知的障害と進行性てんかんを主症状とする男児からPDZドメイン結合モチーフを含むC末端領域の欠損を引き起こすナンセンス変異(p.Q1227*)を見いだした.そして,その変異型BRAG1のシナプスへの局在が障害されることから,BRAG1の神経機能におけるシナプス局在の重要性を明らかにしている29).一方,BRAG3は足場タンパク質であるゲフィリンやジストロフィンとの相互作用により抑制性のGABA作動性シナプス後膜に限局して局在する30–32).なお,BRAG1–3はいずれもArf6に対するGEF活性を有している28, 30, 33, 34).
3)EFA6ファミリー
哺乳類のEFA6(exchange factor for Arf6)またはPSD(PH and Sec7 domain-containing protein)ファミリーは,EFA6A/PSD, EFA6B/PSD4, EFA6C/PSD2, EFA6D/PSD3の四つのメンバーから構成され,Arf6に特異的なGEFとして機能する35–38).EFA6ファミリーはその共通のドメイン構造として,中央にSec7ドメイン,C末端側にPHドメインとCCドメインを持つ.EFA6のC末端領域はアクチン線維と直接結合してアクチン線維を束化し,また隣接するPHドメインと協調して細胞膜とアクチン線維の相互作用を媒介することで,Sec7ドメインを介したArf6の活性化に依存せずに細胞形態に関与する35, 36, 39, 40).EFA6ファミリーのうち,EFA6Bを除く三つは特に神経系に豊富に発現するが,成体マウスの脳におけるその局在は大きく異なる35, 37, 38).EFA6Aは主に嗅球や大脳皮質,海馬,線条体などの前脳領域に豊富に発現しており,海馬CA1錐体神経細胞においては樹状突起およびスパインの細胞膜やエンドソーム,興奮性シナプスのPSD近傍に局在する41, 42).またEFA6Aは,軸索基部にある軸索起始部にも強く濃縮された局在を示す43).EFA6Cは主に小脳プルキンエ細胞に発現しており,プルキンエ細胞の細胞体や樹状突起,スパインの細胞膜や興奮性シナプスのPSDに局在している37).著者らはEFA6Cノックアウトマウスを作製し,EFA6Cの欠損が平行線維–プルキンエ細胞間のシナプス密度の低下を引き起こすことを明らかにしている44).EFA6Dは脳に広く分布しており,海馬CA3錐体神経細胞においては細胞体や樹状突起,スパイン,軸索,さらにはシナプス前終末など,細胞内のさまざまな部位に局在する38, 45).EFA6Dの神経系における機能はまだよくわかっていないが,卵巣がんのバイオマーカーとしての有効性が指摘されている46).また,EFA6Dには複数のスプライスアイソフォームが存在するが45),これらの機能的差異についてもまだ明らかになっていない.
1)樹状突起スパインの形態形成の制御
神経細胞の樹状突起上に形成されるスパインは,アクチン細胞骨格に富む微小な突起で,興奮性シナプスの後部構造である.スパインは発達期に過剰に形成されるが,その後,成熟に伴い不要なスパインが除去されることで,適正な数のスパインが維持されるようになる.発達初期は,運動性の高い細く長い突起(フィロポディア)が形成され,軸索に接触することで頭部の明瞭なマッシュルーム型のスパインへと成熟する.スパインは記憶や学習の基盤として考えられているシナプス可塑性が起こる場であり,スパインの発達・成熟が適切に進行することは高次脳機能においてきわめて重要な現象である.このプロセスの異常は,発達障害や精神神経疾患の病態と密接に関係している47).
Arf6は,発達期と成熟期のどちらにおいてもスパインの形態形成の制御に関わることが海馬や大脳皮質の初代培養系を用いた研究から指摘されている10–12).興味深いことは,Arf6の活性が発達期の神経細胞ではスパイン形成を促進する一方,成熟期では過剰なスパイン形成を抑制するというように,発達段階に応じて異なる方向に制御するという点である.このArf6によるスパインの形成制御機構において,その上流を担うArf6-GEFとしてEFA6Aが着目されており,特に発達期のおける詳細な機構が明らかにされている.Choiらは,発達期の神経細胞においてEFA6AがArf6の活性化を介してフィロポディアをスパインに成熟させることを見いだした10).フィロポディアの細胞膜には接着性膜分子であるテレンセファリン(ICAM5)が豊富に局在しており,これがフィロポディアからスパインへの成熟を阻害している48).そのため,フィロポディアからスパインへの成熟にはテレンセファリンがフィロポディア膜上から除去される必要があるが,この過程にEFA6A-Arf6シグナルによるテレンセファリンのエンドサイトーシスが重要な役割を果たしている49).また,EFA6AのC末端にあるPHおよびCCドメインがArf6活性非依存的にフィロポディア形成を促進する10).これらのことから,EFA6AはArf6非依存的なフィロポディア形成の促進と,このフィロポディアをArf6活性依存的にスパインへと成熟させる働きをしていると考えられる.このように,Arf6およびEFA6Aがスパイン形態形成に果たす役割が培養神経細胞レベルで明らかになってきたが,その一方でこの機構が生体脳でも実際に機能しているのか,そして高次脳機能にどのような役割を果たしているのかは未解明であった.
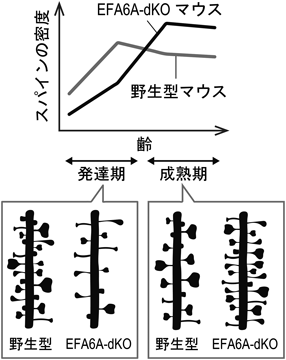
このような背景から著者らは,EFA6A遺伝子から産生される二つのスプライスアイソフォームであるEFA6AとEFA6As(図2)の両方の発現を消失させたEFA6Aダブルノックアウト(EFA6A-dKO)マウスを作製し,発達段階に沿ったスパインの形態形成の推移および海馬の高次機能への影響を調べた.野生型マウスの海馬CA1領域の錐体神経細胞では,発達期においてスパイン密度が大幅に増加しピークに達する.その後,成熟期になるとスパイン密度が低下する(図3).これは,上述した発達期から成熟期にかけてのスパイン形成の推移をよく表した結果である.ところが,EFA6A-dKOマウスでは,発達期までは野生型マウスに比べて有意にスパイン密度が低下していた一方で,成熟期はむしろスパイン密度が顕著に増加していた(図3).成熟期で観察されるスパイン密度の増加はより月齢の進んだマウスにおいても認められたことから,EFA6A-dKOマウスでは単にスパインの発達が遅れたことによりこのような異常が生じたわけではないことが示唆された.つまり,EFA6Aは発達期にはスパイン形成を促進的に,一方で成熟期では抑制的に制御するという,発達段階に応じた双方向性のスパインの制御を行っていることがわかった.また,EFA6A-dKOマウスでは細長い未成熟な形態のスパインが多く観察された.これらの結果は,Arf6活性が発達段階に応じた双方向性のスパイン形態形成の制御に関わること11),またEFA6A-Arf6シグナルがスパインの成熟や維持を制御するという報告と一致していた10, 49).海馬の発達に伴うEFA6Aタンパク質の発現量を調べた結果,EFA6Aは生後7日齢をピークに成熟後はそれよりも減少していた.しかし,成熟後のEFA6A-dKOマウスの海馬では,定常状態のArf6活性が40%以下にまで低下していた.したがって,成熟海馬においてEFA6Aは主要なArf6-GEFであり,EFA6Aの欠損によるArf6活性の低下が発達段階ごとのスパイン形態形成の異常の一要因である可能性が考えられる.一方で,もう一つのスプライスアイソフォームであるEFA6Asは,発達に伴い発現量が増加し,成熟後の海馬ではEFA6Aよりも豊富に発現する主要なアイソフォームであることがわかった.EFA6Asにも共通するC末端領域の構造はArf6非依存的にアクチン細胞骨格を制御する36).これらのことから,EFA6A-dKOマウスで観察されたスパイン形態の異常は,EFA6AとEFA6Asの両方の欠失による複合的な影響であることが示唆される.つまり,EFA6Aによる発達段階に応じたスパイン形成の制御には,EFA6Aのスプライスアイソフォームの発現量のバランスが大きく関わっている可能性が考えられた.さらに,EFA6A-dKOマウスの海馬では,シャーファー側枝–CA1シナプス間の長期増強現象(long-term potentiation:LTP)が消失しており,また受動的回避学習試験で評価される海馬依存的な学習機能の低下もみられた.したがって,EFA6Aは発達期や成熟期といったライフステージごとにスパインの数を適切に調節することで,海馬の機能的な神経回路網の構築と維持に寄与しており,この機構が海馬の機能である記憶学習に重要な役割を果たしていることが個体レベルで初めて明らかになった50).
また,BRAG2の欠損が成熟した大脳皮質の錐体細胞においてスパイン密度の増加とスパインの未成熟化を引き起こすことが報告されている51).これは,Arf6-GEFによるArf6の活性化がスパインの過剰形成の抑制と成熟化に寄与するという著者らの知見と一致している.以上より,スパインの形成・維持には複数のArf6-GEFが関与し,複雑なArf6の活性制御機構が存在することが示唆される.したがって,上流のシグナルによるArf6-GEFの使い分けや,Arf6-GEF自身の活性制御機構のさらなる解明が待たれる.
2)シナプス可塑性における役割
シナプス可塑性は,神経活動に応じて神経伝達効率が可逆的に変化する現象であり,学習や記憶などの高次脳機能の基盤となる細胞レベルでのメカニズムとして考えられている.このシナプス可塑性の主要な機序としては,興奮性シナプス伝達において中心的な役割を担うイオン透過型グルタミン酸受容体の一つであるAMPA型グルタミン酸受容体(AMPAR)のシナプス後膜における量的および質的な変化が考えられている.AMPARの増加によりシナプス強度が持続的に上昇するLTPが引き起こされるのに対し,AMPARの除去によりシナプス強度が低下する長期抑圧現象(long-term depression:LTD)が生じる.シナプス後膜におけるAMPARの発現量は,エキソサイトーシスによる膜への挿入やエンドサイトーシスによる膜からの除去,膜上での側方拡散,リサイクリング経路などの一連の膜輸送系により制御される52, 53).
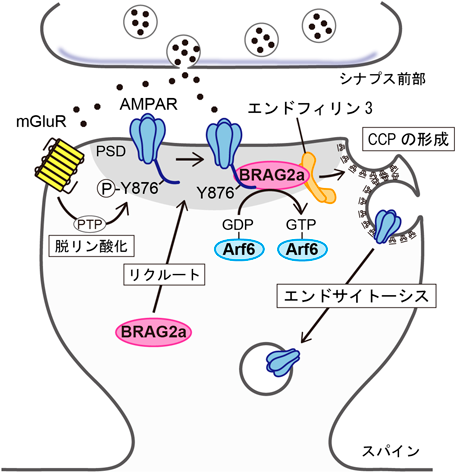
シナプス可塑性の中でも,特にLTDにおけるArf6シグナルの関与がよく知られている.Arf6-GEFの一つであるBRAG2は,AMPARのGluA2サブユニットのC末端領域に結合する13).この結合は,mGluR刺激によって誘導されるGluA2サブユニットのアミノ酸876番目のチロシン残基(Tyr876)の脱リン酸化により引き起こされる.さらに,AMPARへのリガンド結合が同時に起こることで,BRAG2によるArf6の活性化が誘導される.そして,このArf6の活性化を介してAMPARのエンドサイトーシスが引き起こされるという,mGluR依存性のLTDにおけるBRAG2-Arf6シグナルを介した一連の分子機構が明らかにされている13).加えて,著者らはBRAG2のスプライスアイソフォームのうち興奮性シナプス後膜に選択的に局在するBRAG2a(図2)について,C末端領域を餌とした酵母ツーハイブリッドスクリーニングを実施し,BRAG2aがそのC末端領域に存在するプロリンリッチドメインを介してエンドフィリン3と結合することを見いだした26).エンドフィリン3はアダプター複合体AP-2,クラスリン,ダイナミンなどとともにクラスリン依存性エンドサイトーシスを仲介する構成要素であり,AMPARのエンドサイトーシスに関与することが知られている.さらに著者らは,海馬神経細胞において,mGluR1/5の刺激によってBRAG2aとエンドフィリン3との複合体形成が促進されるとともに,Arf6活性が亢進すること,また,BRAG2aとエンドフィリン3の分子間相互作用がmGluR依存的なAMPARのエンドサイトーシスに必要であることを明らかにした26).LTDを誘導したときに起こるクラスリン依存性のエンドサイトーシスでは,細胞膜上のホスファチジルイノシトール4,5-ビスリン酸(PI(4,5)P2)の産生が必須である.Arf6はホスファチジルイノシトール4-リン酸5-キナーゼ(PIP5K)を活性化することでPI(4,5)P2の産生を促し,AP-2をシナプス膜にリクルートすることにより54),クラスリンを集積させクラスリン被覆ピットを形成させる55).したがって,mGluR依存的なLTDにおいて,BRAG2aはAMPARと結合し,その近傍でArf6を活性化するだけでなく,エンドフィリン3をリクルートすることでArf6の活性化をさらに増幅させる.これにより,AP-2やクラスリンなどの集積が促進され,積荷タンパク質であるAMPARをエンドサイトーシス経路へと効率的かつ正確に誘導するプラットフォームとして機能していると考えられる.そして,この機構により,神経活動に応じた受容体のエンドサイトーシスにおける選択性と特異性が確保されていることが示唆された(図4).
上記のmGluR依存的LTDにおけるBRAG2aの役割が明らかになった一方で,ScholzらはNMDA受容体(NMDAR)依存的なLTDにおいてもBRAG2aが必須であることを明らかにしているが13),そのメカニズムは不明である.NMDAR依存性LTDではGluA2-Tyr876の脱リン酸化は必要とされず,むしろリン酸化が必要であることが指摘されていることから56),BRAG2a-Arf6経路がNMDARの刺激に応じたAMPARのエンドサイトーシスを制御するためには,前述とは異なるメカニズムが存在すると考えられる.この点においては,BRAG1に関する報告が重要な手がかりとなる.BRAG1も前述のBRAG2aと同様に,そのPDZドメイン結合モチーフを介してPSD-95と結合することで興奮性シナプス後膜に選択的に局在している.さらに,BRAG1はArf6の活性化を介して海馬神経細胞におけるNMDAR依存的なLTDの発現を制御することが報告されている57, 58).BRAG1のIQモチーフは,カルシウム非結合型のカルモジュリン(アポカルモジュリン)と結合する.NMDARを介した大きなカルシウム流入により,カルシウム結合型カルモジュリンはBRAG1から解離し,BRAG1の可逆的な構造変化が生じ,Arf6が活性化される.このArf6の活性化がc-Jun N末端キナーゼ(JNK)を介したAMPARのエンドサイトーシスを促進する58).また,IQモチーフへのカルモジュリンの結合/非結合による構造変化は,IQモチーフとSec7–PHドメイン間の分子内結合による自己抑制に関与しており,これはBRAGファミリー分子に共通する特徴である59).知的障害を有する家系においてBRAG1のIQモチーフ内に変異が同定されており,これによりカルシウム依存的なBRAG1の活性制御機構が破綻し,Arf6の過剰な活性化が引き起こされることが報告されている58, 59).これらの知見から,mGluR依存的なLTDの機構とは異なり,NMDAR依存的LTDにはBRAG1(おそらくはBRAG2も)のカルシウム依存的な自己活性制御機構が重要であると考えられる.
一方で,Arf6のLTPへの関与はいまだ不明であり,少なくともBRAG1はLTPの発現には関与しない57).しかしながら,前述のEFA6A-dKOマウスにおいてLTPの発現が障害されることが見出されており50),今後,EFA6AによるLTP制御機構の詳細な解明が待たれる.
3)その他の神経機能における役割
サイトへジンファミリーの神経機能として,サイトへジン-2がArf6の活性化を介してArf6のエフェクター分子であるPIP5KやホスホリパーゼDなどのリン脂質修飾酵素を活性化することで海馬神経細胞の軸索の伸長や分岐を抑制的に制御することが報告されている8).また,サイトへジン-1および2は海馬神経細胞の樹状突起の伸長や分岐の制御にも関わる7, 21).さらに著者らは,中枢神経特異的なサイトへジン-2遺伝子欠損マウスを用いて,サイトへジン-2とArf6を介したシグナル経路が慢性疼痛の発症に関わることを明らかにしている22).
BRAG1はマウス海馬神経細胞において軸索の伸長や樹状突起の分岐形成,スパインの形成の制御に関与する51, 60).BRAG2はBRAG1や3とは異なり脳以外にも全身の組織に広く分布しており34),BRAG2の遺伝子欠損マウスは重篤な発達遅延を起こし,胚性致死となる61).BRAG1および2は,先天性知的障害の患者で遺伝子変異が見つかっており,その病態への関与を明らかにするための研究が精力的に行われている29, 51, 62).また,BRAG3はGABA作動性の抑制性シナプスの形成や維持に関与し,その欠損マウスはてんかん発作や不安用行動を引き起こすことが知られている32, 63).さらにBRAG3も先天性知的障害の原因遺伝子である可能性が指摘されている.
EFA6Aはその神経機能として,神経細胞の樹状突起形成64)やスパインの形成および維持10, 49),軸索内逆行性輸送を介した軸索再生の制御43)などに関与する.また興味深いことに,EFA6AにはSec7ドメインを持たずにC末端側のPHドメインとCCドメインのみから構成される短いスプライスアイソフォーム(EFA6As)があり(図2),この短鎖アイソフォームは神経突起の形成において全長のEFA6Aとは異なる作用を示すことが報告されている65).
Arf6-GEFは分子ごとに異なる発現様式,ドメイン構造,GEF活性の特異性,およびGEF活性の制御機構を有している.これらの多様性がArf6-GEFの細胞内局在や相互作用分子,Arf6活性能などの差異をもたらし,Arf6が担う神経機能の多彩さの基盤となっていると考えられる.現在までに,Arf6-GEFとArf6を介したシグナル経路によるさまざまな神経機能の制御機構が明らかにされつつあるが,今後解決すべき課題も残されている.
本稿で述べたように,NMDAR依存性LTDにおいてはBRAG1のカルシウム依存的な活性制御機構が明らかにされている.しかし,ほかの現象やほかのGEFにおいては,どのような刺激や上流シグナルによってGEF活性が制御されるのか,詳細な解明が待たれる.また,同一の細胞内部位に複数のGEFが局在する場合,それらがどのように使い分けられ,またArf6の活性状態にどのような違いが生じるのかという点も興味深い疑問である.さらに,Arf6活性に依存しないArf6-GEFの作用がいくつかのケースで確認されているが,これがArf6活性依存的な作用とどのような関係性を持つのかについても,今後の理解が求められる.
また,本稿では十分にふれることができなかったが,近年,Arf6シグナルとさまざまな精神神経疾患との関連が報告されている.たとえば,BRAGファミリーの遺伝子変異を有する知的障害の患者は,てんかんや自閉症を高率に合併することが報告されている29, 51, 62, 66).さらに,脆弱X症候群のモデルマウスにおけるArf6活性の異常や67),Arf6がアミロイド前駆タンパク質やBACE1の小胞輸送を介してβアミロイドの産生に関与することも明らかになっている68).今後,これらの現象について特に個体レベルでの解析が進展すれば,関連疾患の病態解明,治療法の確立,そして創薬に大きく貢献することが期待される.