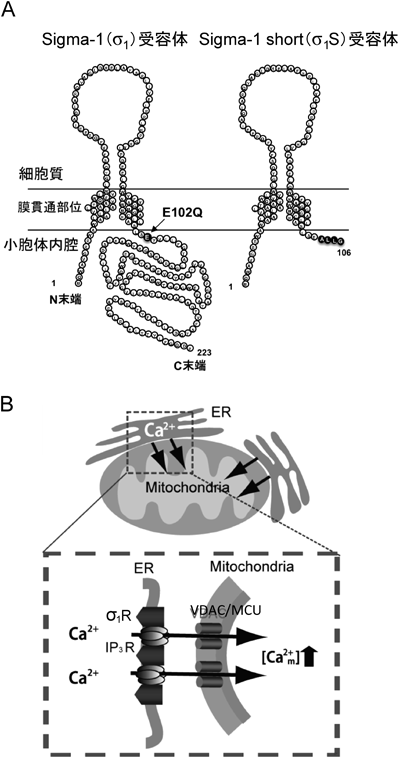
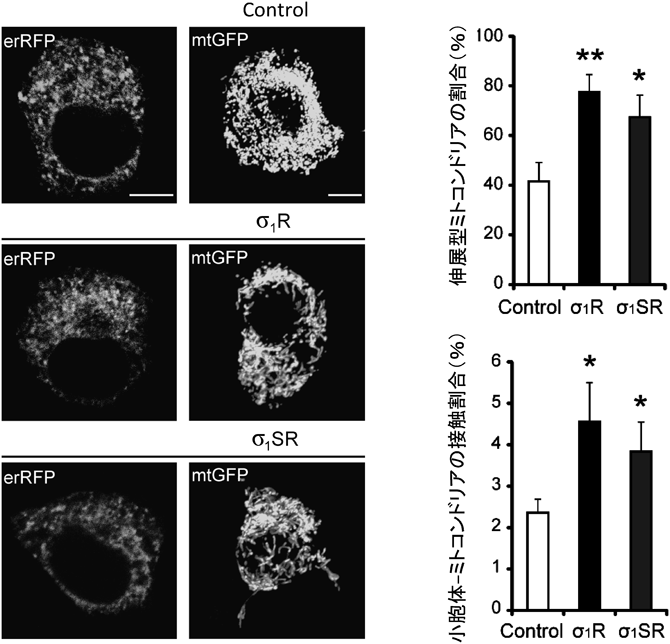
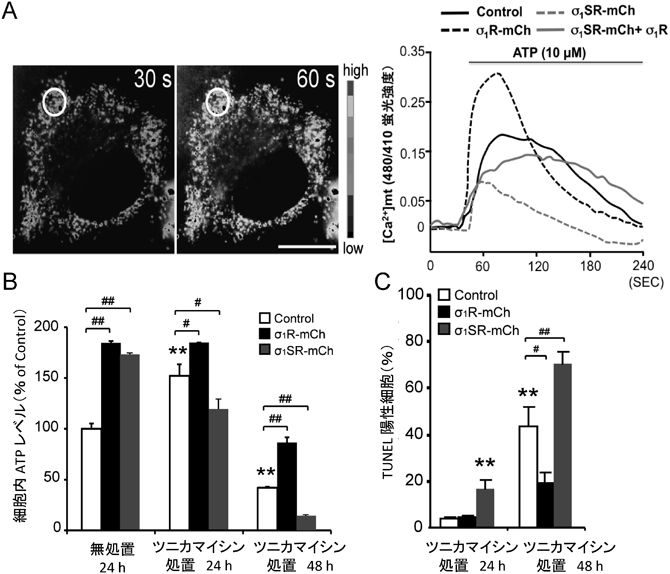
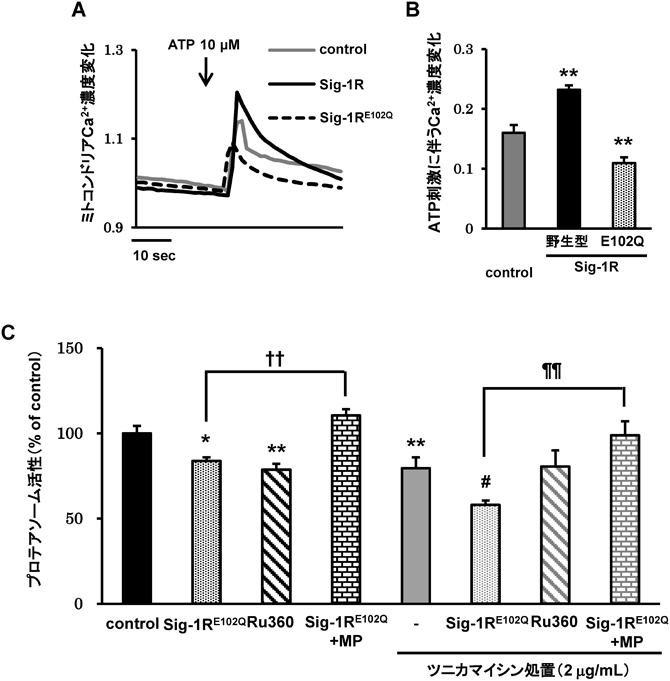
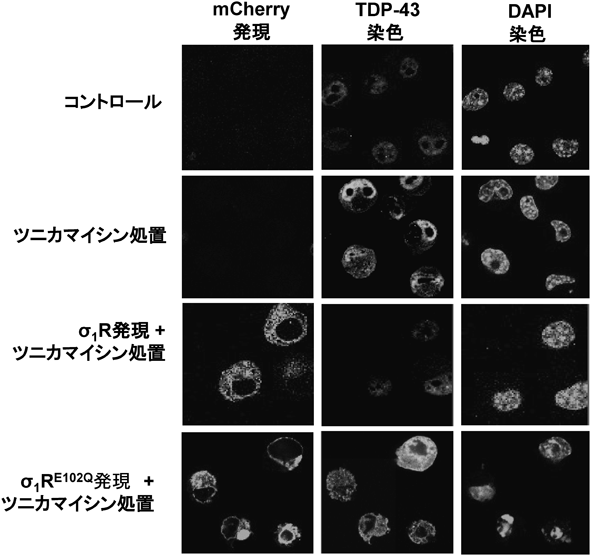
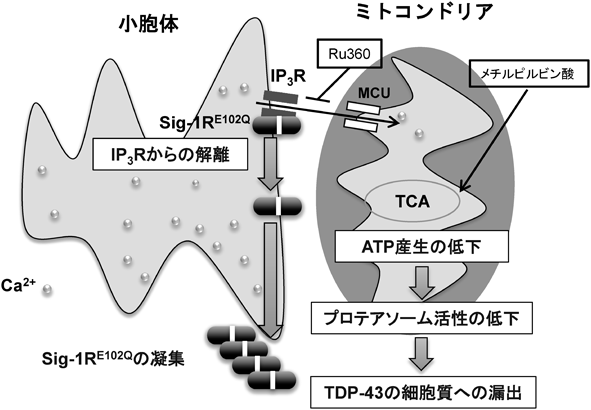
Sigma-1受容体の生理機能と筋萎縮性側索硬化症との関連Pathophysiological role of sigma-1 receptor in Amyotrophic lateral sclerosis
東北大学大学院薬学研究科薬理学分野Department of Pharmacology, Graduate School of Pharmaceutical Sciences, Tohoku University ◇ 〒980–8578 宮城県仙台市青葉区荒巻字青葉6–3 ◇ 6–3 Aramaki-Aoba Aoba-ku, Sendai 980–8578, Japan
発行日:2017年2月25日Published: February 25, 2017