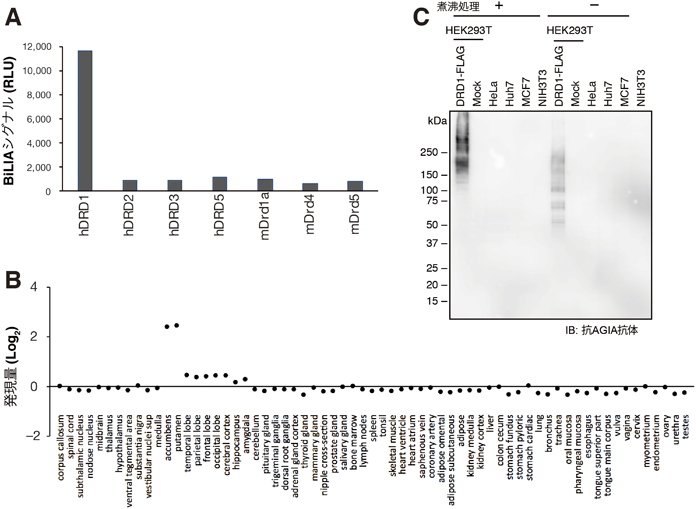
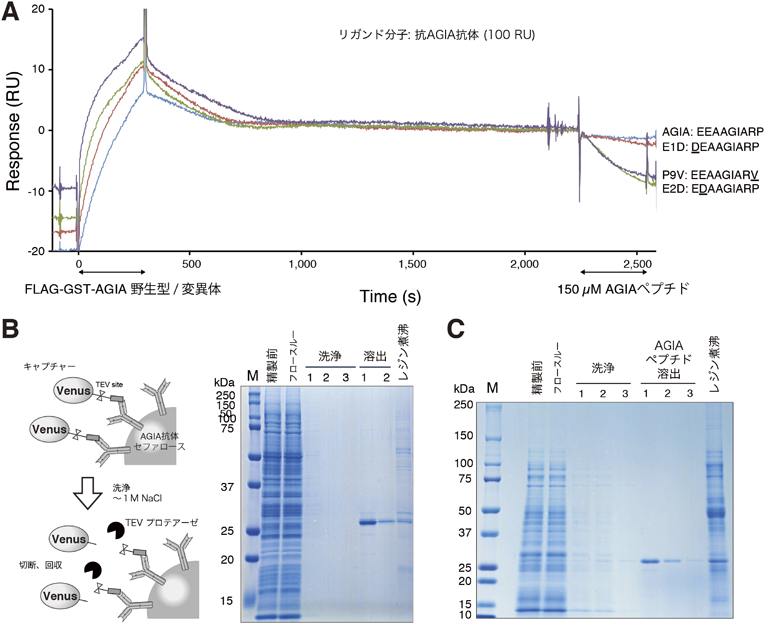
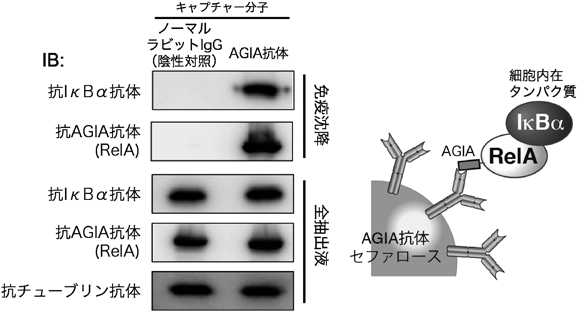
AGIAタグシステム:細胞生物学研究に最適な高感度検出およびキャプチャー用ペプチドタグAGIA tag system: a super-sensitive detection and capture peptide tag suitable for cell biology
愛媛大学プロテオサイエンスセンターProteo-Science Center, Ehime University ◇ 〒790–8577 愛媛県松山市文京町3 ◇ 3 Bunkyo-cho, Matsuyama, Ehime 790–8577, Japan
発行日:2017年4月25日Published: April 25, 2017