1)脂肪酸β酸化の基質
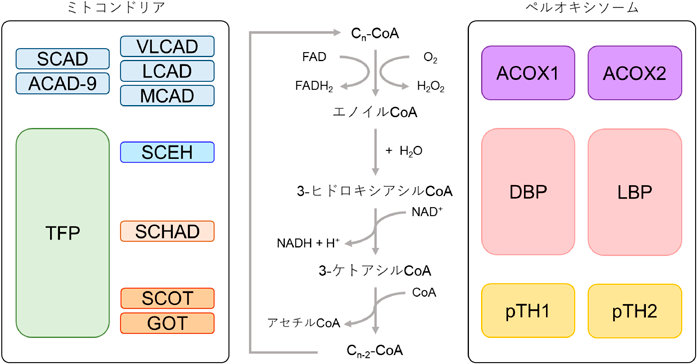
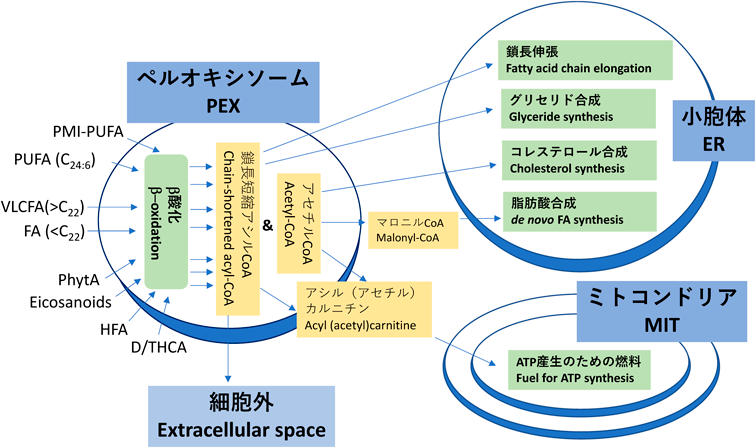
高等生物の細胞における脂肪酸β酸化は,ミトコンドリアとペルオキシソームという二つのオルガネラによって実行されている.図1に示すように,脂肪酸をβ酸化して鎖長短縮するという働きとしては両オルガネラで共通であり,いずれにおいても脂肪酸がカルボン酸側から2炭素分ずつ短縮されていく.しかし,それぞれで関与する酵素はまったく異なる.また,ミトコンドリアにおける脂肪酸β酸化の過程で得られるアセチルCoAやFADH2およびNADHはATP産生,すなわちエネルギー産生に利用されるのに対し,ペルオキシソームではアセチルCoA, NADHおよび過酸化水素が産生され,それらは直接ATP産生に利用されることはない.ミトコンドリアでは短鎖(C4)から長鎖(C20程度)の脂肪酸がβ酸化されるのに対して,ペルオキシソームではC22以上の極長鎖脂肪酸やさまざまな脂溶性カルボン酸性物質もβ酸化の基質として認識される.以下にペルオキシソームのβ酸化基質をあげ,図2にそのβ酸化物の行方を示す.
a.極長鎖脂肪酸
C22以上の脂肪酸,いわゆる極長鎖脂肪酸はもっぱらペルオキシソームでβ酸化される.この事実はZellweger症候群などのペルオキシソーム形成異常症患者やX-ALD, ACOX1欠損症,DBP欠損症のようなペルオキシソームでの脂肪酸β酸化に関与するタンパク質を欠損している患者が,C24やC26といった極長鎖脂肪酸を顕著に蓄積することからも明らかである.
b.2-メチル分枝脂肪酸(プリスタン酸)
後述のように,C20のフィタン酸はα酸化されてC19のプリスタン酸へと変換される.このプリスタン酸はさらにペルオキシソームで3サイクルのβ酸化を受け,排出される8).
c.胆汁酸前駆体(ジ/トリヒドロキシコレスタン酸)
コール酸やケノデオキシコール酸は高等動物における代表的な一次胆汁酸であり,これらはいずれもコレステロールから合成される.まず,コレステロールに種々のシトクロームP450が作用することで,中間体のジ/トリヒドロキシコレスタン酸(D/THCA)が産生される.このD/THCAはペルオキシソームに輸送され,25位のメチル基がα-メチルアシルCoAラセマーゼ(α-methylacyl-CoA racemase:AMACR)によってRからS配置へと変換される.そしてペルオキシソームのβ酸化1サイクルを受け,それぞれが対応する一次胆汁酸へと変換される9).
d.テトラコサヘキサエン酸
ドコサヘキサエン酸(DHA, C22:6, Δ-4, 7, 10, 13, 16, 19)は脳や網膜などに多く存在する多価不飽和脂肪酸(polyunsaturated fatty acid:PUFA)で,これが欠乏することにより脳の発達障害,記憶や視覚に異常を来す.DHAは,α-リノレン酸(C18:3, Δ-9, 12, 15)の伸長および不飽和化によって産生されたドコサペンタエン酸にΔ4不飽和化酵素が作用することで生合成されると考えられてきた.しかし,高等動物においてΔ4不飽和化酵素の発現が認められないことから別の生合成経路の存在が予想された.現在,DHAはテトラコサヘキサエン酸(C24:6, Δ-6, 9, 12, 15, 18, 21)がペルオキシソームのβ酸化1サイクルを受けて鎖長短縮されることで生合成されることが明らかとなっている10).また,ACOX1欠損症患者由来線維芽細胞ではDHA含量が健常人のおよそ半分程度まで減少する11)ことから,細胞中DHAの約50%がペルオキシソームのβ酸化を介して供給されると考えられる.
e.ジカルボン酸
ジカルボン酸は,生体内での脂肪酸のω酸化や,二重結合部分の酸化的開裂に伴って生成する.長鎖ジカルボン酸はもっぱらペルオキシソームでβ酸化され,ミトコンドリアでは代謝されない.ペルオキシソームにおいてジカルボン酸の分解を担う酵素の実態は不明であったが,2012年,L-二頭酵素(L-bifunctional protein:LBP)をコードする遺伝子EHHADH(enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase)ノックアウトマウスの解析から,LBPがジカルボン酸のβ酸化の第2, 3ステップを担うことが,Wandersらによって明らかとされた12).
f.アラキドン酸代謝物
膜リン脂質からホスホリパーゼA2によって切り出されたアラキドン酸は,シクロオキシゲナーゼやリポキシゲナーゼなどの酵素によってさまざまな生理活性脂質へと変換される.シクロオキシゲナーゼが作用した下流で生成するプロスタグランジン13, 14)やトロンボキサン類15),リポキシゲナーゼによって産生されるロイコトリエン類16, 17)やヒドロキシエイコサテトラエン酸(hydroxyeicosatetraenoic acid:HETE)18)もペルオキシソームのβ酸化の基質として認識される.これらは1または2サイクルのβ酸化により鎖長短縮され,尿中へと排泄される.
g.腸内細菌が産生するヒドロキシ脂肪酸
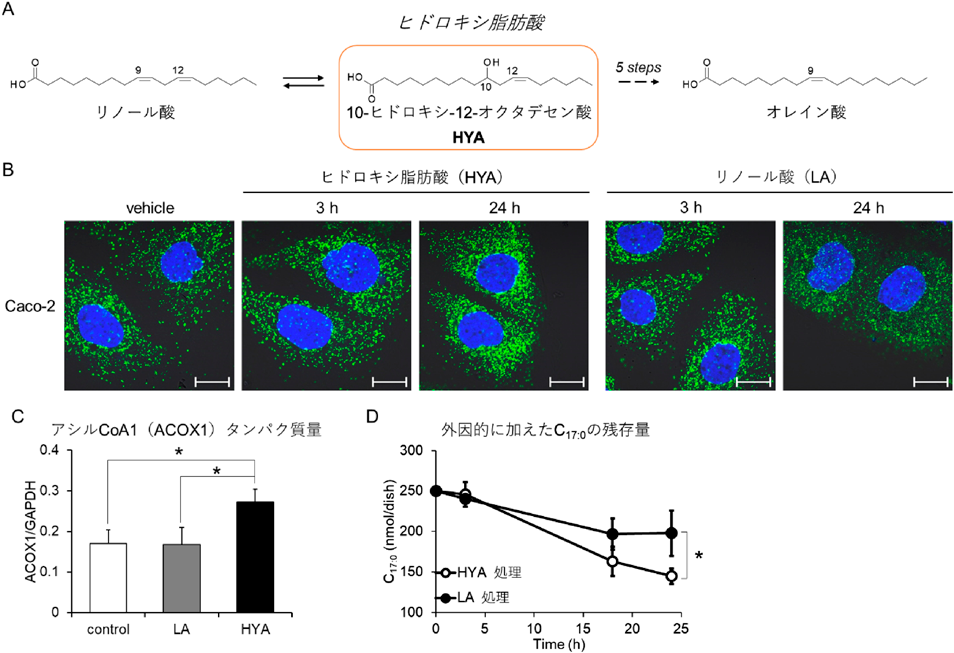
腸内細菌はリノール酸などの二重結合を単結合へと変換,すなわち飽和化することが知られているが,この変換の過程で二重結合部分が水和されたヒドロキシ脂肪酸(図3A)を産生する19).著者らは最近,チャイニーズハムスター卵巣細胞の野生型(CHO-K1)とそのペルオキシソーム欠損型(CHO-zp102)を用いた検討から,腸内細菌が産生するヒドロキシ脂肪酸がペルオキシソームで1~2サイクルのβ酸化を受け,その一部が細胞外へ放出されることを明らかとした.この代謝系はヒト消化管由来の株化細胞においても観察され,我々ヒトにおいてもヒドロキシ脂肪酸の代謝はペルオキシソームが担っていると考えられた20).このとき,添加したヒドロキシ脂肪酸の約50%が24時間で培養系から消失したが,驚くべきことに,このヒドロキシ脂肪酸消失量は細胞の総脂肪酸量とほぼ同じであった.一方,同様の実験をリノール酸を用いて行うと,細胞内への取り込み量はヒドロキシ脂肪酸と同程度だが,そのほとんどがトリアシルグリセロールとして貯蔵されており,培養系からの消失はほとんどなかった.すなわち,ヒドロキシ脂肪酸の代謝様式はリノール酸のそれとは明らかに異なっており,両者の違いは水酸基のみであることから,細胞内ではこの違いを特異的に見分ける機構が存在していると思われる.現在までに,このメカニズムの詳細については明らかとなっていないが,一つの可能性としては,脂肪酸結合タンパク質(fatty acid-binding protein:FABP)への親和性の違いがあげられる.たとえば肝臓型FABP(L-FABP, FABP1とも呼ばれる)は,アラキドン酸よりもそのリポキシゲナーゼ代謝物のヒドロペルオキシエイコサテトラエン酸(hydroperoxyeicosatetraenoic acid:HPETE)やHETEへの結合能が高いことが報告されている21).興味深いことに,L-FABPはペルオキシソームのβ酸化に関与することが示唆されている22).
2)脂肪酸α酸化の基質
脂肪酸を1炭素分鎖長短縮する代謝経路をα酸化と呼び,動物細胞ではペルオキシソームで実行されている.脂肪酸のα酸化は未解明の点も残されているが,現状,1)脂肪酸のα炭素の水酸化,2)2-ヒドロキシアシルCoAリアーゼ(2-hydroxyacyl-CoA lyase 1:HACL1)によるカルボキシ基とα炭素間での切断とそれに伴う長鎖アルデヒドの生成,そして3)長鎖アルデヒド脱水素酵素(ALDH3A2)による長鎖アルデヒドの酸化,のように進行すると考えられている6).以下にα酸化によって代謝される基質をあげる.
a.3-メチル分枝脂肪酸(フィタン酸)
C20のフィタン酸は植物油脂や反芻動物の脂肪および乳製品に含まれる分枝脂肪酸である.このフィタン酸はペルオキシソームのα酸化によってC19のプリスタン酸に変換される.前述のように,このプリスタン酸は引き続きペルオキシソームで3サイクルのβ酸化を受け,排出される.なお,フィタン酸などのα炭素を水酸化するphytanoyl-CoA hydroxylase(PHYH)をコードする遺伝子は,ペルオキシソーム病の一つであるレフサム病の原因遺伝子として知られる.
b.2-ヒドロキシ脂肪酸
ヒトを含む動物や植物,酵母などでは脂肪酸の2位を水酸化する酵素fatty acid 2-hydroxylase(FA2H)が小胞体に発現しており,それによって産生される長鎖から極長鎖の2-ヒドロキシ脂肪酸の多くはスフィンゴ脂質のN-アシル鎖として存在する23).2-ヒドロキシ脂肪酸はペルオキシソームのHACL1とALDH3A2によって1炭素短い(極)長鎖脂肪酸へと代謝される.動物では2-ヒドロキシ脂肪酸は脳や腎臓および皮膚に多く含まれ,これらの組織にはC23やC25といった2-ヒドロキシ脂肪酸が代謝(α酸化後に鎖長伸長)されてできた奇数鎖長の脂肪酸が存在する24).
ペルオキシソームによる脂肪酸β酸化活性は組織や細胞の種類によって異なり,たとえばラット肝細胞の場合,トータルの脂肪酸β酸化活性のうちペルオキシソームに由来するものは5%未満25)から約30%26)までさまざまな数値で報告されている.ただし,いずれにしても,定常状態において一般的な長鎖脂肪酸をβ酸化するのは主にミトコンドリアであり,ペルオキシソームはミトコンドリアの補助的な役割を担う程度に考えられてきた.しかし,ここまで紹介してきたように,ペルオキシソームはミトコンドリアでは酸化消去できない化合物も基質として認識している.また,ミトコンドリアにおける脂肪酸酸化はエネルギー産生の役割が強いのに対し,ペルオキシソームのそれはエネルギー産生と直接は共役していない.つまり,脂肪酸を短縮することそれ自体が目的のように思われる.ここではペルオキシソームにおける脂肪酸酸化の役割について論じていきたい.
1)異物・不要物の分解
前述のように,フィタン酸は我々が普段摂取している食物に含まれており,ペルオキシソームでのα酸化によって分解されている.このα酸化の初発酵素PHYHを遺伝的に欠損する疾患はレフサム病と呼ばれ,全身にフィタン酸を蓄積する.多くの場合,レフサム病患者の血中フィタン酸濃度は200 µM以上を示し,網膜色素変性症や嗅覚・聴覚障害,多発ニューロパチー,心筋症などを呈する.また,各種脂肪酸β酸化酵素の欠損症では極長鎖脂肪酸が蓄積し,多くの場合中枢神経系に異常が現れ,生存期間は長くても10年程度である.すなわち,ペルオキシソームは生体にとっての脂溶性異物,あるいは不要となったものを消去していると考えられる.ペルオキシソーム病患者がいずれも重篤な症状を呈することから,この役割がいかに重要であるかがうかがえる.
著者らは最近,腸内細菌が産生するヒドロキシ脂肪酸がペルオキシソームのβ酸化によって活発に分解されることを示した20).このヒドロキシ脂肪酸も食事や腸管内で産生されることを考慮すれば,異物と捉えられるかもしれない.著者らはこの研究の過程で,ヒドロキシ脂肪酸の代謝が添加後6時間程度から加速するという現象を観察した.これは,ヒドロキシ脂肪酸がペルオキシソーム数の増大および脂肪酸β酸化酵素の誘導作用を示したことに起因した.そして興味深いことに,ヒドロキシ脂肪酸自身のみならず,外因的に加えた脂肪酸の分解活性も亢進させた(図3).古くから,高脂肪食摂取時にペルオキシソーム数および脂肪酸酸化活性の顕著な増大が起こる27)ことが知られていたが,腸内細菌が産生したヒドロキシ脂肪酸もペルオキシソームの数および機能を亢進させ,過剰な脂肪酸を除去するように作用しているのかもしれない.
2)脂質の合成
ペルオキシソームの脂肪酸β酸化は脂質の分解だけでなく生合成にも関与する.D/THCAからの一次胆汁酸産生やテトラコサヘキサエン酸からのDHA産生がそのよい例である.
胆汁酸は食事性に摂取された脂溶性栄養素などをミセル化し,吸収効率を高める役割を果たす.D/THCAをペルオキシソームでβ酸化可能な構造へと変換するAMACRの欠損症患者では胆汁酸欠乏とともに脂溶性ビタミン異常を呈する.その症状としては新生児期からの重度の肝機能異常だけでなく,成人期における感覚運動性ニューロパチーや網膜色素変性症など多岐にわたる.
DHAはエイコサペンタエン酸(EPA, C20:5, Δ-5, 8, 11, 14, 17)とともに魚油に豊富に含まれる脂肪酸であるが,EPAは摂取に伴い血中濃度が上昇するのに対し,DHAはそこまで大きく変化しない28).前述のように,DHAの約50%はペルオキシソームのβ酸化を介して供給されると推測されており11),DHAの体内濃度を維持する上でペルオキシソームが重要な役割を担っていることがわかる.なお,DHAの役割については本特集の守口らの稿に詳しいので参照されたい.
引用文献References
1) De Duve, C. & Baudhuin, P. (1966) Peroxisomes (microbodies and related particles). Physiol. Rev., 46, 323–357.
2) Goldfischer, S., Powers, J.M., Johnson, A.B., Axe, S., Brown, F.R., & Moser, H.W. (1983) Striated adrenocortical cells in cerebro-hepato-renal (Zellweger) syndrome. Virchows Arch. A Pathol. Anat. Histopathol., 401, 355–361.
3) Lazarow, P.B. & de Duve, C. (1976) A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. USA, 73, 2043–2046.
4) Singh, I., Moser, A.E., Goldfischer, S., & Moser, H.W. (1984) Lignoceric acid is oxidized in the peroxisome: implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc. Natl. Acad. Sci. USA, 81, 4203–4207.
5) Heymans, H.S.A., Schutgens, R.B.H., Tan, R., van den Bosch, H., & Borst, P. (1983) Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome). Nature, 306, 69–70.
6) Van Veldhoven, P.P. (2010) Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res., 51, 2863–2895.
7) Wanders, R.J.A. & Waterham, H.R. (2006) Peroxisomal disorders: The single peroxisomal enzyme deficiencies. Biochim. Biophys. Acta, 1763, 1707–1720.
8) Wong, D.A., Bassilian, S., Lim, S., & Lee, W.-N.P. (2004) Coordination of peroxisomal β-oxidation and fatty acid elongation in HepG2 cells. J. Biol. Chem., 279, 41302–41309.
9) Ferdinandusse, S. & Houten, S.M. (2006) Peroxisomes and bile acid biosynthesis. Biochim. Biophys. Acta, 1763, 1427–1440.
10) Voss, A., Reinhart, M., Sankarappa, S., & Sprecher, H. (1991) The metabolism of 7, 10, 13, 16, 19-docosapentaenoic acid to 4, 7, 10, 13, 16, 19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J. Biol. Chem., 266, 19995–20000.
11) Itoyama, A., Honsho, M., Abe, Y., Moser, A., Yoshida, Y., & Fujiki, Y. (2012) Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J. Cell Sci., 125, 589–602.
12) Houten, S.M., Denis, S., Argmann, C.A., Jia, Y., Ferdinandusse, S., Reddy, J.K., & Wanders, R.J.A. (2012) Peroxisomal L-bifunctional Enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J. Lipid Res., 53, 1296–1303.
13) Fauler, J., Tsikas, D., Mayatepek, E., Keppler, D., & Frölich, J.C. (1994) Impaired degradation of prostaglandins and thromboxane in Zellweger syndrome. Pediatr. Res., 36, 449–455.
14) Diczfalusy, U. & Alexson, S.E.H. (1988) Peroxisomal chain-shortening of prostaglandin F2α. J. Lipid Res., 29, 1629–1636.
15) Diczfalusy, U., Vesterqvist, O., Kase, B.F., Lund, E., & Alexson, S.E.H. (1993) Peroxisomal chain-shortening of thromboxane B2: evidence for impaired degradation of thromboxane B2 in Zellweger syndrome. J. Lipid Res., 34, 1107–1113.
16) Jedlitschky, G., Huber, M., Völkl, A., Müller, M., Leier, I., Müller, J., Lehmann, W.D., Fahimi, H.D., & Keppler, D. (1991) Peroxisomal degradation of leukotrienes by β-oxidation from the ω-end. J. Biol. Chem., 266, 24763–24772.
17) Mayatepek, E., Lehmann, W.D., Fauler, J., Tsikas, D., Frölich, J.C., Schutgens, R.B., Wanders, R.J.A., & Keppler, D. (1993) Impaired degradation of leukotrienes in patients with peroxisome deficiency disorders. J. Clin. Invest., 91, 881–888.
18) Gordon, J.A., Figard, P.H., & Spector, A.A. (1990) Hydroxyeicosatetraenoic acid metabolism in cultured human skin fibroblasts. Evidence for peroxisomal β-oxidation. J. Clin. Invest., 85, 1173–1181.
19) Kishino, S., Takeuchi, M., Park, S.-B., Hirata, A., Kitamura, N., Kunisawa, J., Kiyono, H., Iwamoto, R., Isobe, Y., Arita, M., et al. (2013) Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc. Natl. Acad. Sci. USA, 110, 17808–17813.
20) Morito, K., Shimizu, R., Kitamura, N., Park, S.-B., Kishino, S., Ogawa, J., Fukuta, T., Kogure, K., & Tanaka, T. (2019) Gut microbial metabolites of linoleic acid are metabolized by accelerated peroxisomal β-oxidation in mammalian cells. Biochim. Biophys. Acta, 1864, 1619–1628.
21) Raza, H., Pongubala, J.R., & Sorof, S. (1989) Specific high affinity binding of lipoxygenase metabolites of arachidonic acid by liver fatty acid binding protein. Biochem. Biophys. Res. Commun., 161, 448–455.
22) Antonenkov, V.D., Sormunen, R.T., Ohlmeier, S., Amery, L., Fransen, M., Mannaerts, G.P., & Hiltunen, J.K. (2006) Localization of a portion of the liver isoform of fatty-acid-binding protein (L-FABP) to peroxisomes. Biochem. J., 394, 475–484.
23) Alderson, N.L., Rembiesa, B.M., Walla, M.D., Bielawska, A., Bielawski, J., & Hama, H. (2004) The human FA2H gene encodes a fatty acid 2-hydroxylase. J. Biol. Chem., 279, 48562–48568.
24) Kishimoto, Y., Akanuma, H., & Singh, I. (1979) Fatty acid α-hydroxylation and its relation to myelination. Mol. Cell. Biochem., 28, 93–105.
25) Thomas, J., Debeer, L.J., De Schepper, P.J., & Mannaerts, G.P. (1980) Factors influencing palmitoyl-CoA oxidation by rat liver peroxisomal fractions. Substrate concentration, organelle integrity and ATP. Biochem. J., 190, 485–494.
26) Kondrup, J. & Lazarow, P.B. (1985) Flux of palmitate through the peroxisomal and mitochondrial β-oxidation systems in isolated rat hepatocytes. Biochim. Biophys. Acta, 835, 147–153.
27) Ishii, H., Fukumori, N., Horie, S., & Suga, T. (1980) Effects of fat content in the diet on hepatic peroxisomes of the rat. Biochim. Biophys. Acta, 617, 1–11.
28) Schuchardt, J.P., Schneider, I., Meyer, H., Neubronner, J., von Schacky, C., & Hahn, A. (2011) Incorporation of EPA and DHA into plasma phospholipids in response to different omega-3 fatty acid formulations—A comparative bioavailability study of fish oil vs. krill oil. Lipids Health Dis., 10, 145.
29) Sugiura, A., Mattie, S., Prudent, J., & McBride, H.M. (2017) Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature, 542, 251–254.
30) Fan, J., Li, X., Issop, L., Culty, M., & Papadopoulos, V. (2016) ACBD2/ECI2-mediated peroxisome-mitochondria interaction in Leydig cell steroid biosynthesis. Mol. Endocrinol., 30, 763–782.
31) Houten, S.M., Wanders, R.J.A., & Ranea-Robles, P. (2020) Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta, 1866, 165720.
32) Reszko, A.E., Kasumov, T., David, F., Jobbins, K.A., Thomas, K.R., Hoppel, C.L., Brunengraber, H., & Rosiers, C.D. (2004) Peroxisomal fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in rat heart. J. Biol. Chem., 279, 19574–19579.
33) Costello, J.L., Castro, I.G., Hacker, C., Schrader, T.A., Mets, J., Zeuschner, D., Azadi, A.S., Godinho, L.F., Costina, V., Findeisen, P., et al. (2017) ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol., 216, 331–342.
34) Costello, J.L., Castro, I.G., Schrader, T.A., Islinger, M., & Schrader, M. (2017) Peroxisomal ACBD4 interacts with VAPB and promotes ER-peroxisome associations. Cell Cycle, 16, 1039–1045.
35) Wolff, R.L., Christie, W.W., Pédrono, F., & Marpeau, A.M. (1999) Arachidonic, eicosapentaenoic, and biosynthetically related fatty acids in the seed lipids from a primitive gymnosperm, Agathis robusta. Lipids, 34, 1083–1097.
36) Tanaka, T., Morishige, J., Iwawaki, D., Fukuhara, T., Hamamura, N., Hirano, K., Osumi, T., & Satouchi, K. (2007) Metabolic pathway that produces essential fatty acids from polymethylene-interrupted polyunsaturated fatty acids in animal cells. FEBS J., 274, 2728–2737.
37) Tanaka, T., Uozumi, S., Morito, K., Osumi, T., & Tokumura, A. (2014) Metabolic conversion of C20 polymethylene-interrupted polyunsaturated fatty acids to essential fatty acids. Lipids, 49, 423–429.